Suchergebnis
vom: 12.02.2018
Paul-Ehrlich-Institut Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel
BAnz AT 26.03.2018 B5
Paul-Ehrlich-Institut
Bundesinstitut
für Impfstoffe und biomedizinische Arzneimittel
Bekanntmachung
Richtlinie
zur Gewinnung von Spenderhornhäuten
und zum Führen einer Augenhornhautbank
Die vom Vorstand der Bundesärztekammer in seiner Sitzung vom 25. August 2017 auf Empfehlung des Wissenschaftlichen Beirats beschlossene Richtlinie wird nachstehend bekannt gegeben (Anlage).
Paul-Ehrlich-Institut
Bundesinstitut
für Impfstoffe und biomedizinische Arzneimittel
Richtlinie
zur Gewinnung von Spenderhornhäuten
und zum Führen einer Augenhornhautbank
Erste Fortschreibung
gemäß § 16b Absatz 1 Satz 3 des Transplantationsgesetzes
1 Einleitung
Die vorliegende Richtlinie zur Gewinnung von Spenderhornhäuten und zum Führen einer Augenhornhautbank (nachfolgend als Richtlinie bezeichnet) vereint untergesetzliche Regularien der Bundesärztekammer sowie der Sektion Gewebetransplantation und Biotechnologie der Deutschen Ophthalmologischen Gesellschaft. Sie ersetzt damit die Arbeitsrichtlinien (Gute Fachliche Praxis für Hornhautbanken) der Sektion Gewebetransplantation und Biotechnologie der Deutschen Ophthalmologischen Gesellschaft sowie die Richtlinien der Bundesärztekammer zum Führen einer Augenhornhautbank und führt deren Inhalte in einem Regelwerk zusammen.
Mit dem Gesetz über Qualität und Sicherheit von menschlichen Geweben und Zellen (Gewebegesetz) vom 20. Juli 2007 zur Umsetzung der EU-Geweberichtlinie 2004/23/EG und der Durchführungsrichtlinien 2006/17/EG und 2006/86/EG wurden die wesentlichen Regelungen für den Umgang mit menschlichem Gewebe, das zur Anwendung beim Menschen bestimmt ist, im Transplantationsgesetz (TPG), in der TPG-Gewebeverordnung (TPG-GewV), im Arzneimittelgesetz (AMG) und in der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) festgeschrieben (siehe Nummer 17.1 der Bezugsdokumente).
Die Bundesärztekammer wurde gemäß § 16b Absatz 1 TPG ermächtigt, im Einvernehmen mit dem Paul-Ehrlich-Institut als zuständiger Bundesoberbehörde ergänzend zu den Vorschriften der Rechtsverordnung gemäß § 16a TPG (TPG-GewV) den allgemein anerkannten Stand der Erkenntnisse der medizinischen Wissenschaft zur Entnahme von Geweben und deren Übertragung in Richtlinien festzustellen. Das umfasst insbesondere die Anforderungen an die ärztliche Beurteilung der medizinischen Eignung als Gewebespender, die Untersuchung der Gewebespender und die Entnahme, Übertragung und Anwendung von menschlichen Geweben.
Die Einhaltung des allgemein anerkannten Standes der Erkenntnisse der medizinischen Wissenschaft wird vermutet, wenn diese Richtlinie beachtet wird (§ 16b Absatz 2 TPG). Somit gilt die im begründeten Einzelfall widerlegbare Vermutung, dass bei Beachtung der Richtlinie der Stand der wissenschaftlichen Erkenntnisse eingehalten worden ist.
Hinweise in dieser Richtlinie zu den rechtlichen Grundlagen zur Entnahme und Übertragung von menschlichen Augenhornhäuten erfolgen auszugsweise und dienen der ergänzenden Erläuterung. Die Beachtung dieser Richtlinie entbindet nicht von der Pflicht zur Einhaltung der gesetzlichen und verordnungsrechtlichen Vorgaben. Die rechtlichen Grundlagen sind bindend und unterliegen nicht der Vermutungsregelung des § 16b Absatz 2 TPG.
Diese Richtlinie stellt den allgemein anerkannten Stand der Erkenntnisse der medizinischen Wissenschaft vom 31. März 2017 fest und basiert auf den zu diesem Zeitpunkt geltenden rechtlichen Regelungen.
2 Geltungsbereich der Richtlinie
Die vorliegende Richtlinie soll den betroffenen Fachkreisen die notwendige Handlungsgrundlage geben und die erforderlichen Voraussetzungen beschreiben, damit die Gewinnung und Herstellung von Augenhornhäuten unter vergleichbaren Bedingungen durchgeführt wird und hierbei sowie bei der Anwendung von Augenhornhäuten zur Transplantation die höchstmögliche Sicherheit für den Empfänger gewährleistet wird.
Diese Richtlinie gilt für alle Ärztinnen und Ärzte*, die mit
- –
-
dem Gewinnen, den Laboruntersuchungen, der Be- oder Verarbeitung, Konservierung, Lagerung oder dem Inverkehrbringen von Augenhornhäuten bzw. Augen zur Gewinnung von Hornhauttransplantaten und
- –
-
der Anwendung von Augenhornhäuten zur Transplantation
befasst sind.
Soweit für die Durchführung bestimmter Leistungen andere Personen verantwortlich sind, haben auch diese die Richtlinie zu beachten, denn sie stellt gemäß § 16b Absatz 1 TPG den allgemein anerkannten Stand der Erkenntnisse der medizinischen Wissenschaft fest.
Die für diesen Bereich geltenden Gesetze, Verordnungen, Richtlinien sowie Leitlinien und Empfehlungen sind in Nummer 17.1 der Bezugsdokumente aufgeführt. Die von dieser Richtlinie Betroffenen sind gehalten, regelmäßig die Entwicklung dieser Richtlinie sowie der zugrunde liegenden gesetzlichen und untergesetzlichen Regelungen zu beachten und ihre Tätigkeiten im Anschluss an Aktualisierungen zeitnah in Einklang zu bringen.
Die Richtlinie gilt gemäß § 1 Absatz 2 TPG nicht für die Gewinnung, Laboruntersuchung, Be- oder Verarbeitung, Konservierung, Lagerung oder das Inverkehrbringen von Augenhornhäuten, die nicht zur Anwendung beim Menschen bestimmt sind.
3 Spende und Entnahme
3.1 Voraussetzungen für die Entnahme nach dem TPG
Im TPG werden die Voraussetzungen für die postmortale Spende einer Augenhornhaut geregelt. Gemäß den §§ 3 und 4 TPG dürfen Augenhornhäute nur von einem Spender gewonnen werden, der zu Lebzeiten eine Einwilligung zur Gewebespende erteilt hat oder dessen Angehöriger nach seinem Tod im Sinne des Verstorbenen der Gewebespende zugestimmt hat. Darüber hinaus muss der Tod des Gewebespenders der Richtlinie der Bundesärztekammer zur Feststellung des irreversiblen Hirnfunktionsausfalls (siehe Nummer 17.2 des Literaturverzeichnisses) entsprechend festgestellt werden. Dabei genügt die Feststellung durch einen Arzt, wenn der endgültige, nicht behebbare Stillstand von Herz und Kreislauf eingetreten ist und seitdem mehr als drei Stunden vergangen sind. Dies ist im Entnahmebericht entsprechend zu dokumentieren, beispielsweise durch eine Kopie des Leichenschauscheins. Liegen weniger als drei Stunden zwischen irreversiblem Herz-Kreislaufstillstand und geplanter Hornhautentnahme, beispielsweise bei Multiorganspendern, muss die Todesfeststellung gemäß § 3 Absatz 1 Satz 1 Nummer 2 und Absatz 2 Nummer 2 TPG durch zwei dafür qualifizierte Ärzte erfolgen, die den Gewebespender unabhängig voneinander untersucht haben. Die an den Untersuchungen zur Todesfeststellung beteiligten Ärzte dürfen gemäß § 5 Absatz 2 TPG weder an der Entnahme noch an der Übertragung der Gewebe des Spenders beteiligt sein und nicht Weisungen eines Arztes unterstehen, der an diesen Maßnahmen beteiligt ist.
3.2 Anforderungen an den Betrieb von Augenhornhautbanken, an die Gewinnung von Spenderhornhäuten und an die für diese Gewinnung erforderlichen Laboruntersuchungen nach dem AMG
Augenhornhäute können postmortal im Rahmen einer Organspende aber auch unabhängig von einer Organspende gewonnen werden. Die Spendertestung sowie die Gewinnung, Konservierung und Abgabe von Augenhornhauttransplantaten müssen in Zusammenarbeit mit einer Augenhornhautbank erfolgen, die über eine Erlaubnis zur Gewinnung von Gewebe bzw. zur Gewinnung von Augenhornhauttransplantaten gemäß § 20b AMG sowie eine Erlaubnis für die Be- oder Verarbeitung, Konservierung, Prüfung, Lagerung oder das Inverkehrbringen von Augenhornhauttransplantaten gemäß § 20c AMG verfügt. Ebenso muss die Augenhornhautbank über eine Erlaubnis für die Laboruntersuchungen verfügen oder für diese Untersuchungen ein Labor beauftragen, welches eine solche Erlaubnis besitzt. Wird ein Transplantat in den Verkehr gebracht, muss eine Genehmigung nach § 21a Absatz 1 AMG oder beim Verbringen aus einem EU-Land eine Bescheinigung gemäß § 21a Absatz 9 AMG des Paul-Ehrlich-Instituts als zuständiger Bundesoberbehörde gemäß § 77 Absatz 2 AMG vorliegen. Es ist darauf zu achten, dass die nach der AMWHV vorgelegten Unterlagen inhaltlich mit den Genehmigungs-/Bescheinigungsunterlagen beim Paul-Ehrlich-Institut übereinstimmen. Änderungen in den Angaben und Unterlagen sind dem Paul-Ehrlich-Institut gemäß § 21a Absatz 7 AMG mitzuteilen bzw. von diesem zu genehmigen.
Sofern Gewinnung, Be- und Verarbeitung, Konservierung und Abgabe von Augenhornhauttransplantaten unter derselben Verfügungsgewalt stattfinden, d. h. im Rahmen des Betriebs einer lokalen Augenhornhautbank in einer Klinik oder Abteilung einer Einrichtung der medizinischen Versorgung, in der die Transplantate auch angewendet werden, sind für Gewinnung von Gewebe nur eine Erlaubnis der zuständigen Landesbehörde nach § 20b AMG sowie eine Erlaubnis der zuständigen Behörde des Landes, in dem die Betriebsstätte liegt oder liegen soll, im Benehmen mit der zuständigen Bundesoberbehörde nach § 20c AMG für die Be- und Verarbeitung erforderlich. In diesem Fall ist eine Genehmigung nach § 21a AMG nicht erforderlich, denn hier liegen kein Wechsel der Verfügungsgewalt und kein Inverkehrbringen vor.
Gemäß § 20d AMG (sog. „Einhandregelung“) bedarf es keiner Erlaubnis nach § 20b Absatz 1 und § 20c Absatz 1 AMG für eine Person, die Arzt […] ist und die dort genannten Tätigkeiten mit Ausnahme des Inverkehrbringens ausübt, um das Gewebe oder die Gewebezubereitung persönlich bei ihren Patienten anzuwenden; das setzt allerdings voraus, dass z. B. auch die infektionsserologischen Untersuchungen gemäß § 20b Absatz 1 AMG oder die Prüfung gemäß § 20c AMG vom Anwender persönlich durchgeführt werden. Diese Ausnahme gilt nicht für Arzneimittel, die zur klinischen Prüfung bestimmt sind. Die Anzeigepflicht gemäß § 67 AMG ist einzuhalten.
3.3 Anforderungen an die medizinische Eignung des Spenders und an die Laboruntersuchungen, Spenderakte
Augenhornhäute werden in der Regel unabhängig von einer Organspende entnommen, seltener im Rahmen von postmortalen Organspenden. Augenhornhäute dürfen entnommen werden, wenn:
- 1.
-
gemäß § 3 Absatz 1 Satz 1 Nummer 1 TPG der Gewebespender in die Gewebeentnahme eingewilligt hatte (beispielsweise dokumentiert durch einen Organ-/Gewebespendeausweis), oder gemäß § 4 TPG die nächsten Angehörigen der Gewebeentnahme zugestimmt haben. Hierzu müssen die zustimmenden Angehörigen in den letzten zwei Jahren vor dem Tod des Gewebespenders in persönlichem Kontakt mit diesem gestanden haben. Dies muss durch Befragung des nächsten Angehörigen festgestellt werden. Nach § 4 Absatz 2 TPG genügt die Entscheidung des zuerst erreichten Angehörigen, wenn der nächste Angehörige nicht in einer angemessenen Zeit erreichbar ist. Es reicht bei mehreren gleichrangigen Angehörigen aus, wenn von einem die Zustimmung eingeholt wird. Der bekannte Widerspruch eines einzelnen Angehörigen unterbindet die Gewebespende.
- 2.
-
gemäß § 3 Absatz 1 Satz 1 Nummer 2 TPG der Tod des Gewebespenders nach Regeln, die dem Stand der Erkenntnisse der medizinischen Wissenschaft entsprechen, festgestellt ist und
- 3.
-
gemäß § 3 Absatz 1 Satz 1 Nummer 3 TPG der Eingriff durch einen Arzt vorgenommen wird. Die Entnahme von Gewebe darf gemäß § 3 Absatz 1 Satz 2 TPG auch durch andere dafür qualifizierte Personen unter der Verantwortung und nach fachlicher Weisung eines Arztes durchgeführt werden.
Kommt eine Entnahme mehrerer Organe oder Gewebe in Betracht, soll die Einholung der Zustimmung nach § 4 Absatz 1 Satz 3 TPG zusammen erfolgen. Bei der postmortalen Organ- und Gewebeentnahme hat die Organspende gemäß § 9 Absatz 3 TPG gegenüber der Gewebespende Vorrang. Die Entnahme von Geweben bei einem möglichen Spender von Organen nach § 9a Absatz 2 Nummer 1 TPG ist erst dann zulässig, wenn eine von der Koordinierungsstelle beauftragte Person dokumentiert hat, dass die Entnahme oder Übertragung von Organen nicht möglich ist oder durch die Gewebeentnahme nicht beeinträchtigt wird.
Um diese geforderte Einzügigkeit zu gewährleisten, soll die die Einwilligung einholende Person im Rahmen der Einholung der Zustimmung zur Organspende auch nach der Zustimmung zur Gewebespende fragen. Die Entscheidung über den geeigneten Zeitpunkt, den Inhalt und den Umfang sowie die Führung dieses Gesprächs erfordert entsprechende Erfahrung. Die die Einwilligung einholende Person ist diesen Anforderungen gemäß zu qualifizieren. Zu dem Gespräch sollte zur ärztlichen Beurteilung gemäß § 11 Absatz 4 Satz 2 TPG bei potenziellen Organspendern ein Mitarbeiter der Koordinierungsstelle hinzugezogen werden.
3.3.1 Einwilligung, Spenderakte
Das Einholen der Einwilligungserklärung entsprechend der Vorgaben der §§ 3 und 4 TPG erfolgt unter der Verantwortung eines Arztes. Liegt keine Erklärung des Spenders vor und ist auch den nächsten Angehörigen eine solche Erklärung nicht bekannt, so ist gemäß § 4 Absatz 1 Satz 2 TPG die Entnahme nur zulässig, wenn ein Arzt den nächsten Angehörigen über eine in Frage kommende Gewebeentnahme unterrichtet und dieser ihr zugestimmt hat; diese Unterrichtung und Zustimmung sind auch fernmündlich möglich. Gemäß § 4 Absatz 1 Satz 6 TPG kann der nächste Angehörige mit dem Arzt vereinbaren, dass er seine Erklärung innerhalb einer bestimmten vereinbarten Frist widerrufen kann; diese Vereinbarung bedarf der Schriftform.
Gemäß § 4 Absatz 4 TPG hat der Arzt Ablauf, Inhalt und Ergebnis der Beteiligung der nächsten Angehörigen zu dokumentieren und gemäß § 15 Absatz 1 TPG mindestens 30 Jahre aufzubewahren.
Die Feststellung der Spendereignung in der Entnahmeeinrichtung und die für die Gewinnung erforderlichen Laboruntersuchungen in dem Gewebespenderlabor sind gemäß § 33 AMWHV in einer Standardarbeitsanweisung festzulegen.
Für jeden Spender ist von der Entnahmeeinrichtung eine Akte mit Angaben gemäß § 5 Absatz 1 TPG-GewV (siehe Nummer 17.1 der Bezugsdokumente) anzulegen und von einem Arzt zu unterzeichnen.
3.3.2 Auswahlkriterien für Hornhautspender
Im Rahmen der Prüfung der Eignung eines Verstorbenen zur Gewebespende sind gemäß § 7 TPG insbesondere Ärzte, Einrichtungen der medizinischen Versorgung und Behörden verpflichtet, unverzüglich Auskunft zu geben. Die Eignung zur Gewebespende ist von einem Arzt nach § 8d Absatz 1 Nummer 2 TPG zu überprüfen.
Gemäß § 3 Absatz 1 TPG-GewV beruht die ärztliche Beurteilung zur medizinischen Eignung des toten Spenders nach § 8d Absatz 1 Satz 2 Nummer 2 TPG auf der Risikobewertung in Bezug auf die jeweilige Verwendung der Spenderaugen bzw. -hornhäute und der Art der Gewebe. Anzeichen für solche Risiken sind gemäß Anlage 1 der TPG-GewV (siehe Nummer 17.1 der Bezugsdokumente) durch Anamnese, zweckdienliche Quellen wie Krankenakten und Befragung behandelnder Ärzte, biologische Untersuchung, postmortale Untersuchung und sonstige geeignete Untersuchungen zu ermitteln. Auch eine körperliche Untersuchung des toten Spenders ist hierzu entsprechend Anlage 1 der TPG-GewV (siehe Nummer 17.1 der Bezugsdokumente) vor der Gewebeentnahme erforderlich. Der Spender ist von der Spende auszuschließen, wenn es Hinweise auf eines der Ausschlusskriterien gibt, die in der Anlage 1 (siehe Nummer 16.1 Anlage 1 der Spenderausschlusskriterien) der gemäß § 3 Absatz 1 TPG-GewV für Augenhornhäute spezifizierten Anlage 1 der TPG-GewV aufgeführt sind. Allerdings sieht § 3 Absatz 1 Satz 3 TPG-GewV die Ausnahmeregelung vor, dass „im Einzelfall aus medizinischen Gründen und aufgrund einer Risikobewertung durch einen Arzt hiervon abgewichen“ werden kann. Die Risikobewertung ist zu dokumentieren.
3.3.3 Laboruntersuchungen auf Infektionsmarker
Bei allen Spendern sind dem allgemein anerkannten Stand der Erkenntnisse der medizinischen Wissenschaft entsprechend und gemäß Anlage 3 der TPG-GewV mindestens folgende biologische Tests durchzuführen:
| a) | HIV 1 und 2 | Anti-HIV 1 und 2 |
| b) | Hepatitis B | HBsAg, Anti HBc |
| c) | Hepatitis C | Anti HCV, HCV-NAT |
| d) | Syphilis | siehe unten |
Ist der Anti-HBc-Test positiv und der HBsAg negativ, so kann eine negative HBV-PCR (Mindestsensitivität 12 IU/ml) die klinische Verwendbarkeit ermöglichen.
Zum Ausschluss einer Infektion mit Treponema pallidum ist ein validierter Testalgorithmus einzusetzen. Ein spezifischer oder nicht spezifischer nicht reaktiver Test kann die Freigabe der Gewebe bzw. Zellen ermöglichen. Wird ein nicht spezifischer Test durchgeführt, steht ein reaktives Untersuchungsergebnis der Entnahme oder Freigabe nicht entgegen, sofern ein spezifischer Test zur Bestätigung von Treponema pallidum nicht reaktiv ist. Ein Spender, dessen Probe auf einen spezifischen Treponema-Test reagiert, ist einer gründlichen Risikobewertung zu unterziehen, um die klinische Verwendbarkeit festzustellen.
HTLV-I-Antikörpertests sind bei Spendern vorzunehmen, die in Gebieten mit hoher Prävalenz leben oder daher stammen oder deren Sexualpartner oder Eltern aus solchen Gebieten stammen.
Unter bestimmten Umständen sind zusätzliche Laboruntersuchungen erforderlich, je nach Vorgeschichte des Spenders und den Merkmalen der gespendeten Gewebe.
Die Laboruntersuchungen sind in einem qualifizierten Labor durchzuführen, welches folgende Bedingungen erfüllt:
- 1.
-
Das Labor verfügt über eine eigene Erlaubnis nach § 20b Absatz 1 AMG und hat eine Vereinbarung mit einer Hornhautbank nach § 9 AMWHV über die Durchführung der jeweiligen Spenderuntersuchungen abgeschlossen oder
- 2.
-
das Labor übt die Tätigkeiten unter vertraglicher Bindung mit einer Hornhautbank, die über eine § 20c-Erlaubnis verfügt, aus. In diesem Fall muss die Hornhautbank auch die Erlaubnis nach § 20b Absatz 2 AMG für die Durchführung der Spenderuntersuchungen besitzen. Bei Spendern, die nicht nur Gewebe sondern auch Organe spenden, ist im Einzelfall zu überprüfen, ob das mit der Durchführung der Untersuchung anlässlich der Organspende beauftragte Labor über eine Erlaubnis gemäß § 20b AMG verfügt. Ist dies nicht der Fall, so müssen die Tests in einem Labor wiederholt werden, das über eine behördliche Erlaubnis gemäß § 20b AMG verfügt.
Die biologischen Tests werden am Serum oder Plasma des Spenders, wobei das verwendete Material genau zu dokumentieren ist, vorgenommen. Sie sollten nicht an anderen Flüssigkeiten oder Sekreten, wie z. B. Humor aqueus oder Humor vitreus, durchgeführt werden, sofern dies nicht eigens durch Verwendung eines für eine solche Flüssigkeit validierten Tests klinisch gerechtfertigt ist.
Bei verstorbenen Spendern müssen gemäß Anlage 3 der TPG-GewV entweder bis zu sieben Tage vor dem Tod entnommene Blutproben verwendet werden oder, falls dies nicht möglich ist, müssen die Blutproben so schnell wie möglich und nicht später als 24 Stunden nach dem Tod entnommen werden. Hierbei ist die Möglichkeit einer Verwechslung von prämortal entnommenen Blutproben (Rückstellproben) zu berücksichtigen und dieses Risiko durch geeignete Maßnahmen zu minimieren (z. B. Kontrolle und Dokumentation der Beschriftung der Probengefäße).
Allgemeine Anforderungen an die Bestimmung biologischer Marker
Die Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen in der jeweils gültigen Fassung ist zu beachten. Die Art des zu verwendenden Tests ist mit Blick auf den Verwendungszweck nach dem Stand der wissenschaftlichen Erkenntnisse zu validieren. Die Testsysteme zur Überprüfung postmortaler Blutproben müssen ebenfalls validiert worden sein, entweder durch den Hersteller oder durch das anwendende Labor. Auf die Validierungsempfehlungen des Paul-Ehrlich-Institutes (siehe Nummer 17.2 des Literaturverzeichnisses) wird hingewiesen.
3.3.4 Verwendung von Spenderhornhäuten für wissenschaftliche Zwecke
Die Verwendung von Hornhautgewebe zur wissenschaftlichen Nutzung ist vom Geltungsbereich des TPG und damit von dieser Richtlinie gemäß § 16b TPG nicht erfasst. Allgemeine Regelungen finden sich u. a. im AMG, in den Datenschutzgesetzen der Länder und des Bundes sowie in Bestattungsgesetzen der Länder, die in ihren Regelungen variieren.
Die Datenschutzgesetze enthalten u. a. Regelungen zur Verarbeitung und Nutzung personenbezogener Daten durch Forschungseinrichtungen, zur Zulässigkeit der Datenerhebung, -verarbeitung und -nutzung sowie zur Einwilligung. Personenbezogene Daten dürfen nur für einen im Voraus bestimmten Zweck erhoben und verwendet werden. Der Zweckbindungsgrundsatz ist eng mit der Frage verknüpft, wie konkret sich die vom Spender erteilte Einwilligung auf die spätere Verwendung des Proben- und Datenmaterials beziehen muss.
Spenderhornhäute oder Teile hiervon sowie nach der Transplantation verbleibendes corneales oder sklerales Restgewebe können den rechtlichen Bestimmungen entsprechend für wissenschaftliche Zwecke verwendet werden. Für in vitro-Studien werden nur Spenderhornhäute verwendet, die den Qualitätsstandards zur Verwendung am Menschen nicht genügen und somit verworfen würden.
Die Spende ist freiwillig. Der Spender hat zu Lebzeiten oder der Totensorgeberechtigte hat nach dessen Tod in die Verwendung von Augenhornhäuten zu Forschungszwecken eingewilligt bzw. ihr zugestimmt.
Das geplante Forschungsvorhaben ist entsprechend den gesetzlichen und berufsrechtlichen Regelungen der zuständigen, nach Landesrecht eingerichteten Ethik-Kommission vorzulegen. Das Ethikvotum ist auch der Entnahmeeinrichtung nach § 20b AMG und der kooperierenden Gewebebank bekannt zu geben.
3.4 Entnahmeverfahren für Spenderhornhäute
Gemäß § 2 TPG-GewV ist die Entnahmeeinrichtung insbesondere verpflichtet, sicherzustellen, dass die Art und Weise der Entnahme hinsichtlich der Art der gespendeten Gewebe geeignet ist und die für ihre Verwendung erforderlichen biologischen und physikalischen Eigenschaften erhalten bleiben. Die Entnahme der Spenderaugen bzw. -hornhäute erfolgt durch gemäß § 34 Absatz 1 AMWHV durch ein Schulungsprogramm qualifizierte Personen, die gemäß § 3 Absatz 1 Satz 1 TPG Ärzte sind oder gemäß § 3 Absatz 1 Satz 2 TPG qualifizierte Personen unter der Verantwortung und nach fachlicher Weisung eines Arztes sind.
Für die Gewinnung von Gewebe durch Entnahmeeinrichtungen gelten die Regelungen des § 34 AMWHV. Die Entnahmeverfahren müssen diejenigen Eigenschaften der Spenderhornhaut schützen, die für deren letztendliche klinische Verwendung erforderlich sind, und gleichzeitig das Risiko einer mikrobiellen Verunreinigung während des Verfahrens minimieren, insbesondere da Spenderhornhäute nicht sterilisiert werden können.
Als Entnahmeorte kommen Räume innerhalb, aber auch außerhalb des Krankenhauses, dem eine Hornhautbank angegliedert ist, in Frage, beispielsweise Institute für Pathologie oder Rechtsmedizin, klinische Stationen oder andere Bereiche von Krankenhäusern sowie Bestattungsinstitute. Bei der Augenhornhautentnahme ist grundsätzlich die Regelung des „mobilen Entnahmeteams“ gemäß § 34 Absatz 2 Nummer 4 AMWHV möglich.
Der Zugang zum Entnahmeort ist zu beschränken. Der Entnahmeort sollte zum Entnahmezeitpunkt nicht gleichzeitig durch andere Personen genutzt werden. Er ist so zu wählen, dass eine offensichtliche Kontamination durch z. B. Schmutzaufwirbelung und Aerosolerzeugung in unmittelbarer Nähe ausgeschlossen wird.
Die gesamte Entnahme der Spenderhornhaut bzw. des Spendergewebes, abgesehen von der Räumlichkeit, erfolgt analog zu einem ophthalmochirurgischen Eingriff an einem lebenden Patienten. Das die Entnahme durchführende Personal muss der Art der Entnahme entsprechend gekleidet sein, um das Risiko einer Kontamination des zu entnehmenden Gewebes, aber auch einer Eigengefährdung zu minimieren. Dazu gehört, sich ordnungsgemäß zu desinfizieren (chirurgische Händedesinfektion), sterile Kleidung sowie sterile Handschuhe, Gesichtsmasken bzw. Schutzmasken und Kopfschutz zu tragen.
Es ist im Entnahmegebiet ein lokaler steriler Bereich mit sterilen Tüchern einzurichten, nachdem eine wirksame Haut- und Bindehautdesinfektion mit einem geeigneten Desinfektionsmittel (z. B. 1 – 10 % PVP-Jod, z. B. 3 Minuten Einwirkzeit, Entfernung des Jods z. B. durch Ausspülen oder -wischen) erfolgt ist.
Wird vor oder nach der Desinfektion von Haut und Bindehaut eine für die Cornea repräsentative Probe für eine mikrobiologische Ausgangsmaterialkontrolle entnommen, unterliegt die mikrobiologische Testung dieser Probe der Erlaubnis nach § 20b oder § 20c AMG. Das Ergebnis dieser Untersuchung ist für die Freigabe der Spenderhornhäute nicht relevant.
Für die Bulbus- und Hornhautentnahme sind sterile Medizinprodukte (Instrumente und Entnahmebestecke) zu verwenden. Die Instrumente und Entnahmebestecke müssen für ihren Verwendungszweck geeignet und qualitativ hochwertig sein und sind regelmäßig für die Entnahme von Geweben instand zu halten. Werden wiederverwendbare Instrumente benutzt, so muss ein validiertes Reinigungs- und Sterilisierungsverfahren zur Entfernung von Infektionserregern vorhanden sein, welches dem zur routinemäßigen Aufbereitung von Operationsinstrumenten nach § 8 der Medizinprodukte-Betreiberverordnung entspricht.
Falls medizinische Geräte (z. B. elektrische Trepansysteme) für die Entnahme verwendet werden, gilt § 34 Absatz 2 Nummer 1 AMWHV und die Anforderungen der Medizinprodukte-Betreiberverordnung sind einzuhalten. Das gesamte beteiligte Personal ist nach Vorgabe der Medizinprodukte-Betreiberverordnung auf geeignete Weise im Umgang mit solchen Geräten zu schulen.
Nach der Gewebeentnahme ist der Leichnam des Gewebespenders gemäß § 6 TPG in würdigem Zustand zur Bestattung zu übergeben. Die Leiche ist so wiederherzustellen, dass sie die größtmögliche Ähnlichkeit mit ihrer ursprünglichen anatomischen Form aufweist. Dies kann, soweit aufgrund der Entnahmetechnik erforderlich, durch Verwendung geeigneter Prothesen sowie den Verschluss der Lider erfolgen.
Bei Leichenspendern ist der Entnahmeort aufzuzeichnen und der zwischen dem Tod und der Entnahme verstrichene Zeitraum anzugeben, um sicherzustellen, dass die erforderlichen biologischen und/oder physikalischen Eigenschaften der Gewebe erhalten bleiben.
Die Gewebe entnehmende Person bzw. Organisation erstellt einen Entnahmebericht gemäß § 34 Absatz 7 AMWHV in Verbindung mit § 5 Absatz 2 TPG-GewV, welcher der Hornhautbank übermittelt wird. Im Entnahmebericht ist zu dokumentieren, dass die Gewebe für die Aufbereitung, Be- oder Verarbeitung, Konservierung oder Aufbewahrung freigegeben sind. Die Angaben des Entnahmeberichts sind in § 5 TPG-GewV und § 34 Absatz 7 AMWHV geregelt (siehe Nummer 17.1 der Bezugsdokumente).
Alle Abweichungen im Prozess sowie während der Entnahme aufgetretene Zwischenfälle sind einschließlich der daraufhin erfolgten Untersuchung zu dokumentieren. Die für die Entnahme verantwortliche Person hat gemäß § 5 Absatz 2 TPG-GewV und § 34 Absatz 7 Satz 5 AMWHV im Entnahmebericht mit Datum und Unterschrift zu bestätigen, dass die Entnahme entsprechend der Entnahmeanweisung durchgeführt worden ist und die Gewebe im Sinne des § 8d Absatz 1 Nummer 4 TPG freigegeben sind.
Nach der Entnahme sind die gewonnenen Spenderhornhäute bzw. -augen so zu verpacken, dass das Kontaminationsrisiko minimiert wird. Verpackung und Transport müssen der Spende angemessen sein und die Eigenschaften der Gewebe schützen, die für ihre Verwendung erforderlich sind, sowie das Risiko einer mikrobiellen Verunreinigung der Spende minimieren. Hierfür eignet sich bei der Corneoskleralkomplexentnahme besonders ein steriler, luftdicht verschließbarer Schraubbehälter aus Kunststoff mit Kulturmedium. Für die Bulbusentnahme sollte ein steriler, luftdicht verschließbarer Schraubbehälter aus Kunststoff mit einer sterilen, physiologischen Elektrolytlösung verwendet werden, der eine „Feuchte Kammer“ erzeugt. Die Spenderhornhäute sind bei Temperaturen zu lagern und zu transportieren, bei denen ihre Merkmale und biologischen Funktionen erhalten bleiben. Diese Temperaturen entsprechen grundsätzlich den unten erwähnten Lagerungstemperaturen, in Abhängigkeit von der Kultivierungsmethode. Der Transport von Spenderaugen sollte gekühlt erfolgen bei Temperaturen zwischen + 1 °C und + 10 °C (z. B. mit Kühlelementen). Temperaturen unter 0 °C sind strikt zu vermeiden. Die Transportdauer ist dabei so zu begrenzen, dass die maximal akzeptable postmortale Zeit (Postmortemzeit + Entnahmedauer + gegebenenfalls Transport bei Bulbi), bis die entnommenen Hornhäute in Kulturmedien gelangen, von 72 Stunden (für Organkultur) bzw. 16 Stunden (für Kurzzeitkultur) nicht überschritten wird. Die Transportdauer von der Entnahmeeinrichtung bis zur Gewebebank sollte 48 Stunden nicht überschreiten.
Die mittels Corneoskleralkomplexentnahme gewonnenen Spenderhornhäute für Organkultivierung können bei Temperaturen von ≥ + 10 °C bis ≤ + 40 °C zur Hornhautbank transportiert werden.
Die Packung und die Art des Transports müssen so gewählt werden, dass die Einhaltung der oben genannten Temperaturen sicher gewährleistet ist. Dies ist durch die Hornhautbank in regelmäßigen Abständen zu überprüfen. Sollte eine Temperaturstabilität durch die Verpackung und die Transportdauer nicht zweifelsfrei gesichert werden können (Transportvalidierung), so ist ein Temperaturmessgerät beizulegen, welches mindestens halbstündlich die Temperatur im Inneren der Verpackung misst und die Daten speichert. Die Verpackung muss außerdem die Kontaminationsrisiken der für die Verpackung und den Transport der Gewebe verantwortlichen Personen minimieren.
Die verpackten Gewebe sind in einem Behälter zu transportieren, der für den Transport biologischen Materials geeignet ist und der die Sicherheit und die Qualität der darin enthaltenen Gewebe gewährleistet.
Etwaige zur Testung bestimmte beigefügte Gewebe- oder Blutproben sind genau zu kennzeichnen, um die Identifizierung des Spenders sicherzustellen, zudem müssen sie Angaben über Ort und Zeit der Probennahme tragen.
3.5 Kennzeichnung der entnommenen Gewebe
Für die Gewinnung von Gewebe durch die Entnahmeeinrichtung gelten die Regelungen des § 34 AMWHV. Jedes Gewebe enthaltende Transportgefäß ist zum Zeitpunkt der Entnahme zu kennzeichnen. Dabei sind die Vorgaben des Single European Code (Spendennummer bzw. Spendenkennungssequenz) zu beachten (siehe Nummer 4.5). Die Gewebespende oder, falls die Angaben nicht auf dem Behältnis gemacht werden können, die Begleitdokumentation muss mindestens Angaben gemäß § 34 Absatz 6 AMWHV tragen (siehe Nummer 17.1 der Bezugsdokumente).
Für den Transport zur Be- oder Verarbeitung und Entgegennahme von Gewebe in der Gewebeeinrichtung gilt § 35 AMWHV. Die Behältnisse für den Transport des Gewebes zur Be- oder Verarbeitung sind mit mindestens Angaben gemäß § 35 Absatz 2 AMWHV (siehe Nummer 17.1 der Bezugsdokumente) zu versehen.
Für die Be- oder Verarbeitung und Lagerung durch die Gewebeeinrichtung gelten die Regelungen des § 36 AMWHV. Demnach müssen Gewebe und Gewebezubereitungen vor ihrem Inverkehrbringen mit den Angaben gemäß § 36 Absatz 8 AMWHV in Verbindung mit § 10 Absatz 8b AMG (siehe Nummer 17.1 der Bezugsdokumente) gekennzeichnet werden.
4 Organisationsstruktur und Qualitätsmanagement
Eine Hornhautbank muss insbesondere gemäß § 8d TPG in Verbindung mit § 32 AMWHV über eine Organisationsstruktur verfügen, die für die Verarbeitung von Spenderhornhäuten geeignet ist. In einem Organigramm müssen die Verantwortlichkeiten und Weisungsbefugnisse eindeutig ersichtlich sein.
4.1 Qualitätsmanagementsystem
Jede Hornhautbank muss gemäß § 32 AMWHV ein dokumentiertes Qualitätsmanagementsystem nach den Grundsätzen der „Guten Fachlichen Praxis“ verwenden und auf dem neuesten Stand halten.
Die Hornhautbanken treffen alle erforderlichen Maßnahmen, um sicherzustellen, dass im Rahmen des Qualitätssicherungssystems für alle Prozesse Standardarbeitsanweisungen gemäß den §§ 32 bis 41d AMWHV vorliegen.
4.2 Dokumentation und Aufzeichnungen
Es muss gemäß § 32 AMWHV ein System für eine klar definierte, korrekte und vollständige Dokumentation vorhanden sein. Das System muss sicherstellen, dass alle Schritte nachvollziehbar sind, d. h. die Kodierung, Spendereignungsprüfung, Gewinnung, Be- oder Verarbeitung, Konservierung, Lagerung, der Transport, das Inverkehrbringen und die Entsorgung. Diese Dokumentation muss von der angemessen ausgebildeten Person nach § 20b AMG bzw. von der verantwortlichen Person nach § 20c AMG regelmäßig auf Aktualität und Effizienz überprüft werden.
Das System muss unter der Aufsicht der verantwortlichen Person stehen und sicherstellen, dass eine Spenderhornhaut den Anforderungen an Sicherheit und Qualität für die Freigabe und Verteilung entspricht. Dies sollte auf einem Prüfprotokoll erfolgen, in dem alle sicherheits- und qualitätsrelevanten Punkte aufgelistet sind.
Für jede kritische Tätigkeit (Entnahme und Präparation, Lagerung, sowie alle Manipulationen an der Spenderhornhaut) sind die entsprechenden Materialien und Ausrüstungen sowie das Personal zu identifizieren und zu dokumentieren.
Die Aufzeichnungen müssen lesbar und unauslöschlich sein. Sie dürfen handgeschrieben sein oder auf ein anderes validiertes System (Computer) übertragen werden.
Alle Aufzeichnungen, die für die Sicherheit und Qualität der Spenderhornhaut kritisch sind, sind so zu führen, dass der Zugang zu diesen Daten gemäß § 15 Absatz 2 TPG und den Anforderungen nach § 41 AMWHV mindestens 30 Jahre nach dem Ablauf des Verfallsdatums des Gewebes, der Übertragung des Gewebes oder der Entsorgung sichergestellt ist. Nach Ablauf der Aufbewahrungsfrist sind die Angaben zu löschen oder zu anonymisieren.
Die Aufzeichnungen müssen den gültigen datenschutzrechtlichen Bestimmungen entsprechen. Der Zugang zu den Daten ist auf Personen, die von der verantwortlichen Person dazu die Befugnis erhalten, sowie die zuständige Behörde zwecks Inspektions- und Kontrollmaßnahmen zu beschränken.
4.3 Personal
Das Personal der Hornhautbank muss gemäß §§ 20b Absatz 1 Satz 3 Nummer 1 und 2, 20c Absatz 2 Satz 1 Nummer 1 und 2 AMG in ausreichender Anzahl vorhanden und für seine Aufgaben qualifiziert sein. Die Kompetenz des Personals ist regelmäßig im Rahmen des Qualitätssystems zu bewerten.
Alle Mitarbeiter müssen über klare, dokumentierte und aktuelle Aufgabenbeschreibungen verfügen. Jeder Hornhautbank muss ein approbierter Arzt zur Verfügung stehen, der über die medizinische Tätigkeit in der Hornhautbank (z. B. Spenderauswahl und Kontraindikationsprüfung, Prüfung klinischer Ergebnisse und serologischer Tests) berät und Aufsicht führt.
Die Meldeverfahren und Schulungsverpflichtungen gemäß §§ 34 und 40 AMWHV sind zu beachten. Das Personal muss einführende und grundlegende Schulungen, bei Bedarf (z. B. Änderungen der Verfahren oder neuen wissenschaftlichen Erkenntnissen) aktualisierte Schulungen erhalten.
Die Schulungsprogramme müssen sicherstellen, dass alle Mitarbeiter sich bei der Durchführung der ihnen zugewiesenen Aufgaben als kompetent erweisen.
Die Mitarbeiter müssen den organisatorischen Rahmen, das Qualitätsmanagementsystem sowie die Gesundheits- und Sicherheitsvorschriften der Einrichtung, in der sie arbeiten, verstehen und ausreichend über den weiteren ethischen, rechtlichen und gesetzlichen Zusammenhang ihrer Arbeit informiert sein.
Jede Hornhautbank benennt eine verantwortliche Person, die mindestens die Voraussetzungen und Qualifikationen gemäß § 20c Absatz 3 AMG erfüllen muss.
Die Hornhautbank hat der zuständigen Landesbehörde den Namen und die Qualifikation der verantwortlichen Person mitzuteilen. Bei einem unvorhergesehenen Wechsel der verantwortlichen Person hat die Hornhautbank gemäß § 20c Absatz 6 AMG der zuständigen Behörde unverzüglich den Namen der neuen verantwortlichen Person und das Datum, an dem sie ihre Tätigkeit aufnimmt, mitzuteilen. Bei einem geplanten Wechsel der verantwortlichen Person darf die Änderung erst vollzogen werden, wenn die zuständige Behörde eine schriftliche Erlaubnis erteilt hat.
4.4 Festzuhaltende Mindestdaten der Spenderhornhaut, des Spenders, des Empfängers
Die in der Hornhautbank gespeicherten Daten müssen Angaben gemäß § 8d Absatz 2 TPG und § 5 TPG-GewV (siehe Nummer 17.1 der Bezugsdokumente) umfassen.
Die Übertragung von Gewebe ist durch Einrichtungen der medizinischen Versorgung gemäß § 13a TPG in Verbindung mit § 7 TPG-GewV zu dokumentieren.
4.5 Kennzeichnung und Rückverfolgbarkeit
Die Hornhautbank hat gemäß den §§ 13a bis 13c TPG in Verbindung mit den §§ 7 bis 9 TPG-GewV über ein wirksames System zu verfügen, mit dem die eingegangenen und verteilten Spenderhornhäute eindeutig identifiziert und gekennzeichnet werden. Dieses muss gewährleisten, dass die Spenderhornhaut und gegebenenfalls alle ihre Teile zu jedem Zeitpunkt eindeutig identifizierbar sind.
Das System zur Identifizierung von Spenderhornhäuten muss auf allen Stufen der Verarbeitung in der Hornhautbank eindeutig freigegebene von nicht freigegebenen (in der Quarantäne befindlichen) und ausgesonderten Spenderhornhäuten unterscheiden. Zum Zeitpunkt der Entnahme müssen Gewebe gemäß § 41b AMWHV mit einer eindeutigen Spendennummer gekennzeichnet werden. Die Entnahmeeinrichtung, die eine eigene § 20b-AMG-Erlaubnis besitzt, oder die Gewebeeinrichtung, die von einer Entnahmeeinrichtung ohne eigene § 20b-AMG-Erlaubnis die Gewebespende erhält, ergänzt die eindeutige Spendernummer mit ihrem EU-Gewebeeinrichtungs-Code gemäß Abschnitt 5b AMWHV zu einer Spendenkennungssequenz.
Es ist sicherzustellen, dass sämtliche Spenderhornhäute vom Spender zum Empfänger und umgekehrt zurückverfolgt werden können. Diese Rückverfolgbarkeit betrifft auch alle einschlägigen Daten über Produkte und Materialien, die mit diesen Geweben in Berührung kommen. Für jede kritische Tätigkeit ist die Identifizierung des ausführenden Personals sicherzustellen.
4.6 Aufbewahrung der Daten und Datenschutz
Es gilt § 15 TPG. Alle Aufzeichnungen müssen im Einklang mit den Datenschutzvorschriften während des gesamten angegebenen Aufbewahrungszeitraums klar erkennbar, leicht abrufbar und vor unbefugter Änderung geschützt sein sowie in diesem Zustand aufbewahrt werden.
Die an der Hornhautentnahme, -bearbeitung und -übertragung beteiligten Personen dürfen personenbezogene Daten der Gewebespender und der Gewebeempfänger nicht offenbaren.
Spätestens nach Eingang der Spenderhornhäute/-augen in der Hornhautbank ist eine Pseudonymisierung (z. B. durch eine Kennnummer) vorzunehmen. Die Lagerung, gegebenenfalls Bearbeitung und Verteilung hat in pseudonymisiertem Zustand zu erfolgen.
Die im Zusammenhang mit der Hornhautspende erhobenen personenbezogenen Daten dürfen ausschließlich für die Bereitstellung von Hornhauttransplantaten verarbeitet oder genutzt werden. Die für eine vollständige Rückverfolgbarkeit erforderlichen Aufzeichnungen gemäß § 15 Absatz 2 TPG und § 41 AMWHV sind mindestens 30 Jahre nach der klinischen Verwendung oder dem Verfallsdatum aufzubewahren.
4.7 Beziehungen zwischen Hornhautbanken und Dritten
Hornhautbanken müssen schriftliche Vereinbarungen mit einem Dritten nach § 9 AMWHV schließen, wenn eine Tätigkeit, z. B. die Entnahme von Gewebe, außerhalb der Einrichtung erfolgt. Die Hornhautbank kann insbesondere in folgenden Fällen Vereinbarungen mit Dritten treffen:
- 1.
-
Übertragung eines Teils der Hornhautgewinnung und/oder -verarbeitung.
- 2.
-
Übertragung von Laboruntersuchungen (Infektionsserologie, mikrobiologische Untersuchungen).
- 3.
-
Übertragung der Verteilung von freigegebenen Spenderhornhäuten.
Die Beurteilung und Auswahl Dritter ist von der Hornhautbank sorgfältig vorzunehmen. Die Hornhautbank führt eine vollständige Liste ihrer mit Dritten abgeschlossenen Vereinbarungen. In den Vereinbarungen zwischen Hornhautbanken und Dritten sind die Verantwortlichkeiten, die von Dritten wahrgenommen werden, und die genauen Verfahren festzulegen. Der Dritte muss die gesetzlichen Anforderungen erfüllen. Die Anforderungen dieser Richtlinie sind einzuhalten, entsprechende Verpflichtungen sind in die Vereinbarung zu übernehmen. Auf Verlangen der zuständigen Behörde(n) legen die Hornhautbanken Kopien ihrer Vereinbarungen mit Dritten vor.
4.8 Register, Jahresbericht und Berichtspflicht der Hornhautbank
Die Hornhautbank führt gemäß § 8d Absatz 3 TPG eine Statistik als Zusammenstellung ihrer Tätigkeiten.
Eine Veröffentlichung zu den Leistungs- und Komplikationszahlen (Jahresbericht jeder Hornhautbank nach von der Sektion Gewebetransplantation und Biotechnologie der Deutschen Ophthalmologischen Gesellschaft vorgegebenem Schema) soll ebenso erfolgen wie ein Jahresbericht der Sektion Gewebetransplantation und Biotechnologie der Deutschen Ophthalmologischen Gesellschaft.
Gemäß § 8d TPG besteht für alle Gewebeeinrichtungen eine gesetzliche Berichtspflicht gegenüber der Bundesbehörde, d. h. dem Paul-Ehrlich-Institut, im Hinblick auf die Zahl der entnommenen, be- und verarbeiteten, abgegebenen/transplantierten, eingeführten und ausgeführten Gewebe. Die TPG-Meldung muss jährlich bis zum 1. März des Folgejahres auf vom Paul-Ehrlich-Institut zur Verfügung gestellten Formblättern erfolgen (siehe Nummer 17.1 der Bezugsdokumente).
5 Einrichtungen und Räumlichkeiten
Eine Hornhautbank muss nach den arzneimittelrechtlichen Vorgaben in AMG und AMWHV über geeignete Einrichtungen verfügen, die dem Hauptzweck entsprechen. Der Betrieb in der Hornhautbank soll vom Klinik- und Patientenbetrieb getrennt sein. Insbesondere ist auf eine strikte Trennung zwischen Verarbeitungsbereich sowie sonstigen Räumen zu achten. Der Verarbeitungsbereich der Hornhautbank ist ausschließlich für den Zweck der Verarbeitung, Konservierung, Beurteilung und Lagerung von humanen Geweben zur Transplantation zu verwenden.
Bei allen Bearbeitungsschritten in der Hornhautbank, bei der die Spenderhornhaut gegenüber der Umgebung exponiert ist, ist während der Be- oder Verarbeitung ein Luftreinheitsgrad für Keimzahl und Partikelzahl entsprechend der Klasse A der Definition des EG-GMP-Leitfadens Anhang 1 (Bekanntmachung vom 12. März 2008, BAnz. S. 1217), mit einer für die Be- oder Verarbeitung des Gewebes geeigneten Hintergrundumgebung, die in Bezug auf Partikel- und Keimzahl mindestens der Klasse D des Anhangs 1 des Leitfadens entspricht, erforderlich.
Die Eignung der Sicherheitswerkbank als Reinraum der Klasse A und die Eignung des umgebenden Raumes als Reinraum der Klasse D ist durch Qualifizierung/regelmäßige Requalifizierung nach den von Anhang 1 des oben genannten EG-GMP-Leitfadens vorgegebenen Kriterien und Methoden nachzuweisen.
Zusätzlich ist die Einhaltung der Umgebungsbedingungen während der Be- und Verarbeitung der Gewebe durch regelmäßige Partikelmessungen und mikrobiologisches Monitoring ebenfalls nach den Vorgaben des Anhangs 1 des EG-GMP-Leitfadens nachzuweisen. Ein Monitoringplan ist zu erstellen.
6 Ausrüstungen und Materialien
Sämtliche Ausrüstungen müssen den arzneimittelrechtlichen Vorgaben in AMG und AMWHV entsprechend ihres Verwendungszweckes gestaltet und gewartet werden. Sie müssen eine Gefährdung der Empfänger und/oder des Personals auf ein Minimum begrenzen.
Alle kritischen Ausrüstungen und technischen Geräte (insbesondere die Laminar-Air-Flow Box, bei der Organkultur die Brutschränke und bei der hypothermen Lagerung die Kühlschränke) sind zu identifizieren, zu qualifizieren und deren Funktion zu überprüfen, regelmäßigen Inspektionen zu unterziehen und nach den Anweisungen des Herstellers vorbeugend zu warten. Sind durch die Ausrüstung kritische Parameter berührt, müssen diese Gegenstände angemessene Überwachungsvorrichtungen besitzen, damit Fehlfunktionen und Defekte festgestellt werden können und damit gewährleistet ist, dass die kritischen Parameter zu jedem Zeitpunkt innerhalb akzeptabler Grenzen gehalten werden. Alle Ausrüstungen mit einer kritischen Messfunktion sind, soweit verfügbar, anhand einer verfolgbaren Norm zu kalibrieren. Dies gilt insbesondere für das Mikroskop (und gegebenenfalls Bildverarbeitungsprogramme) mit dem die Endothelzelldichte der Spenderhornhaut bestimmt wird.
Neue, gewartete und reparierte Ausrüstungen sind bei der Installation zu testen, vor dem Gebrauch zu qualifizieren und durch die verantwortliche Person freizugeben. Die Testergebnisse sind zu dokumentieren.
Wartung, Instandsetzung, Reinigung, Desinfektion und Dekontaminierung aller kritischen Ausrüstungen sind regelmäßig durchzuführen und entsprechend in einem Logbuch zu dokumentieren.
Es müssen Verfahren für den Betrieb jedes Teils der kritischen Ausrüstung vorhanden sein, die ausführlich angeben, welche Maßnahmen im Falle von Fehlfunktionen oder Versagen zu ergreifen sind.
Die Standardarbeitsanweisungen müssen die Spezifikationen aller kritischen Materialien und Reagenzien ausführlich angeben.
7 Qualitätsüberprüfungen
Prüfungen der Gewebe sind gemäß § 37 AMWHV durchzuführen. Abweichungen von den vorgeschriebenen Qualitäts- und Sicherheitsstandards müssen zu dokumentierten Untersuchungen führen, die eine Entscheidung über mögliche Korrektur- und Präventivmaßnahmen einschließen. Was mit nicht vorschriftsmäßigen Spenderhornhäuten geschieht, ist anhand schriftlicher Verfahrensanweisungen unter Aufsicht der verantwortlichen Person zu entscheiden und aufzuzeichnen. Alle betroffenen Spenderhornhäute und gegebenenfalls deren Teile müssen identifiziert und dokumentiert werden.
Korrekturmaßnahmen sind zeitnah und effektiv zu dokumentieren, einzuleiten und abzuschließen. Präventiv- und Korrekturmaßnahmen sind nach der Implementierung auf ihre Wirksamkeit zu überprüfen.
8 Eingang der Spendergewebe in der Hornhautbank
Wenn die entnommenen Spenderhornhäute/-augen in der Hornhautbank ankommen, wird gemäß § 35 AMWHV überprüft und dokumentiert, ob die Lieferung, einschließlich der Transportbedingungen, der Verpackung, der Kennzeichnung sowie der zugehörigen Unterlagen und Blutproben, den Vorschriften und den Spezifikationen der Hornhautbank entspricht.
Jede Hornhautbank muss sicherstellen, dass die Gewebe unter Quarantäne aufbewahrt werden, bis sie zusammen mit den betreffenden Unterlagen auf ihre Vorschriftsmäßigkeit hin kontrolliert oder überprüft worden sind.
Die Hornhautbank muss über dokumentierte Verfahrensanweisungen für den Umgang mit Sendungen verfügen, die den Vorschriften nicht entsprechen, um jegliches Kontaminationsrisiko für andere Gewebe auszuschließen. Die Annahme oder Ablehnung entgegengenommener Gewebe ist zu dokumentieren. Wurde das Gewebe einem Spender entnommen, bei dem auch Organe zum Zwecke der Übertragung entnommen worden sind, unterrichtet die Hornhautbank gemäß § 35 Absatz 3 Satz 5 AMWHV die Koordinierungsstelle nach § 11 Absatz 1 TPG unverzüglich über die Spenderidentität nach § 34 Absatz 7 Satz 2 Nummer 2 AMWHV. Dabei ist anzugeben, ob das entgegengenommene Gewebe angenommen oder abgelehnt worden ist.
9 Kultivierung und Kontrolluntersuchungen
Prinzipiell ist die Kultivierung von Spenderhornhäuten auf zwei Arten möglich. Hierbei handelt es sich um die Organkultur und die Kurzzeitkultur (hypotherme Lagerung).
Das Kulturmedium ist ein wesentlicher Punkt zur Gewährleistung der Qualität und Sicherheit der Spenderhornhaut. Es ist daher ein für den Zweck der Hornhautkonservierung geeignetes Kulturmedium zu verwenden (z. B. MEM mit Serumzusatz).
Der Zusatz von Rinderserum ist unter folgenden Voraussetzungen erlaubt:
- 1.
-
Serum entspricht der Monografie Ph. Eur. 5.2.8. im Hinblick auf die Minimierung eines BSE-Risikos (EDQM/TSE-Zertifikat).
- 2.
-
Test auf Mycoplasmen ist negativ.
- 3.
-
Serum ist sterilfiltriert bzw. durch ein anderes Verfahren sterilisiert worden.
- 4.
-
Serum entspricht der Monografie Ph. Eur. „Bovine serum“ und ist entsprechend virusinaktiviert.
Auch nach Desinfektion des Auges mit geeigneten Desinfektionsmitteln wie PVP-Jodlösung kann nicht von einer Sterilität des Ausgangsmaterials ausgegangen werden. Daher werden dem Kulturmedium üblicherweise Antibiotika und Antimykotika zugesetzt. Die für dieses Verfahren zur Keimreduktion angewendeten Wirkstoffe sind auf der Grundlage der zu erwartenden Bakterien- und Pilzspezies auszuwählen und in definierten Konzentrationen einzusetzen.
Für die Kultivierung von Spenderhornhäuten hält jede Hornhautbank mindestens nachfolgende Kultivierungsbedingungen ein, die notwendig sind, um die erforderlichen biologischen Eigenschaften der Spenderhornhaut aufrechtzuerhalten.
Diese sind für die verschiedenen Kultivierungsmethoden folgende:
- 1.
-
Hypotherme Lagerung/Kurzzeitkultur im geschlossenen System bei normaler Raumluft: Temperatur zwischen + 2 °C und + 8 °C.
- 2.
-
Organkultur im geschlossenen System: Temperatur zwischen + 30 °C und + 38 °C bei normaler Raumluft. Zur Vorbereitung der Transplantation (nur für vordere lamelläre und perforierende Keratoplastik, nicht zwingend für hintere lamelläre Keratoplastik) anschließend: Organkultur in geeignetem Entquellungs-/Transportmedium in geschlossenem System (Temperatur zwischen ≥ + 10 °C und ≤ + 40 °C bei normaler Raumluft).
- 3.
-
Organkultur im offenen System (mit Begasung): Temperatur zwischen + 30 °C und + 38 °C bei normaler Raumluft mit 5 % +/– 1 % CO2-Anreicherung. Zur Vorbereitung der Transplantation (nur für vordere lamelläre und perforierende Keratoplastik, nicht zwingend für hintere lamelläre Keratoplastik) anschließend: Organkultur in geeignetem Entquellungs-/Transportmedium im offenen System (mit Begasung; Temperatur zwischen ≥ + 10 °C und ≤ + 40 °C bei normaler Raumluft mit 5 % +/– 1 % CO2-Anreicherung).
Unabhängig von der Kultivierungsmethode gilt: Das Vorgehen bezüglich der lamellären Keratoplastik erfordert eine gesonderte Genehmigung nach § 21a AMG, sofern die lamelläre Präparation unter der Verantwortung der Hornhautbank zum Zweck des Inverkehrsbringens vorgenommen wird. Wird eine lamelläre Präparation nach dem Inverkehrbringen unter der Verantwortung des anwendenden Arztes für die Transplantation hergestellt, ist keine Genehmigung nach § 21a AMG erforderlich.
Entsprechend des initialen Volumens des Kulturmediums werden folgende Zeiten für den Mediumwechsel bei organkultivierten Augenhornhäuten definiert:
| Initiales Volumen des Kulturmediums | Zeitpunkt des Medienwechsels |
|---|---|
| 40 bis 60 ml | 3. bis 10. Tag |
| 60 bis 100 ml | 3. bis 14. Tag |
| 100 ml | 3. bis 28. Tag |
Die Hornhautbank stellt sicher, dass sämtliche Lagerungsprozesse unter kontrollierten Bedingungen stattfinden.
Es sind Lagerungseinrichtungen zu verwenden, die die Spenderhornhäute vor der Freigabe bzw. in Quarantäne eindeutig von denjenigen trennen und unterscheiden, die freigegeben wurden, sowie von denjenigen, die verworfen werden, um Verwechslungen zu vermeiden. Alle Gewebe und Gewebezubereitungen müssen vor ihrem Inverkehrbringen gekennzeichnet werden (siehe Nummer 12.2 und 12.3).
Für jede Art der Lagerungsbedingungen ist die Höchstdauer zu spezifizieren. Der gewählte Zeitraum muss unter anderem die mögliche Verschlechterung der erforderlichen Zell- und Gewebeeigenschaften berücksichtigen.
- 1.
-
Für die Organkultur gilt dabei ein Zeitraum von insgesamt 34 Tagen, einschließlich der Lagerung im geeigneten Entquellungs-/Transportmedium, wobei diese sechs Tage nicht überschreiten sollte.
- 2.
-
Für die hypotherme Lagerung beträgt die Höchstdauer 14 Tage.
Die kritischen Verfahrensschritte sind daraufhin zu validieren, dass die Funktionalität und die Sicherheit der Gewebe gewährleistet sind, d. h. die Spenderhornhäute oder Teile hiervon dürfen nicht klinisch unwirksam oder schädlich für den Empfänger sein. Die Validierung kann auf Studien beruhen, welche die Hornhautbank selbst durchgeführt hat, oder auf Daten aus Veröffentlichungen, sofern vergleichbare Bedingungen vorliegen, oder, bei etablierten Verarbeitungsverfahren, auf einer nachträglichen Bewertung der klinischen Ergebnisse.
Die Hornhautbank nimmt sämtliche Verarbeitungsschritte und Kontrolltests, die die Qualität und die Sicherheit berühren, in ihre Standardarbeitsanweisungen auf und stellt sicher, dass sie unter kontrollierten Bedingungen und in Übereinstimmung mit der „Guten Fachlichen Praxis“ durchgeführt werden.
Es ist nachzuweisen, dass die validierten Verfahren vom Personal in der Hornhautbank einheitlich gemäß den genehmigten Standardarbeitsanweisungen durchgeführt werden (z. B. in einem Herstellungsprotokoll).
Vor der Implementierung einer signifikanten Änderung der Verarbeitung ist das geänderte Verfahren zu validieren und zu dokumentieren. Das Kultivierungsverfahren ist regelmäßig kritisch zu bewerten, um sicherzustellen, dass es weiterhin die angestrebten Ergebnisse erzielt.
Die Verfahren zur Aussonderung von Spenderhornhäuten müssen die Kontamination anderen Spendergewebes, der Verarbeitungsumgebung und des Personals vermeiden.
Kontrolluntersuchungen
Das Kulturmedium ist regelmäßig makroskopisch auf Anzeichen einer Kontamination zu überprüfen. Hierbei ist insbesondere auf Trübung oder Änderung der Indikatorfarbe des Mediums sowie auf ungewöhnliche Ablagerungen in der Flasche und das Aussehen der Corneoskleralscheibe zu achten. Besteht der Verdacht einer Kontamination, so ist eine geeignete mikrobiologische Untersuchung zu veranlassen und die betroffenen Spenderhornhäute unter erweiterte Quarantäne zu stellen.
Während der Kultivierung muss gemäß § 37 AMWHV mindestens einmal eine Prüfung von Gewebe und Gewebezubereitungen auf Einhaltung der festgelegten Spezifikation nach vorher erstellter Standardarbeitsanweisung (Prüfeinweisung) in Übereinstimmung mit der „Guten Fachlichen Praxis“ durchgeführt werden. § 33 Absatz 3 AMWHV gilt entsprechend.
Während der Kultivierung ist eine mikroskopische Untersuchung der Endothelzellschicht durchzuführen. Dies sollte bei Spenderhornhäuten (nur für vordere lamelläre und perforierende Keratoplastik, nicht zwingend für hintere lamelläre Keratoplastik) im entquollenen Zustand, kurz vor der geplanten Transplantation stattfinden. Dabei ist die Gesamtendothelfläche zu begutachten und die zentrale Endothelzelldichte durch eine standardisierte und etablierte Zählmethode zu ermitteln.
Aufgrund des zusätzlichen Be- und Verarbeitungsschrittes ist im Falle der Präparation des hinteren lamellären Transplantats und anschließender erneuter Lagerung durch die Augenhornhautbank eine weitere mikrobiologische Kontrolluntersuchung des Mediums durchzuführen, in dem sich das lamelläre Hornhauttransplantat befindet. Für die Beurteilung kann ebenfalls das „Negative-to-date“-Konzept mit Ablesung eines Zwischenergebnisses angewendet werden. Das Zwischenergebnis der mikrobiologischen Kontrolluntersuchung ist freigaberelevant.
Im Rahmen der Kultivierung der Spenderhornhaut ist eine zuverlässige mikroskopische Untersuchung an der Spaltlampe zum Ausschluss von sichtbaren, pathologischen Veränderungen durchzuführen.
Sowohl während der Organkultur als auch der hypothermen Lagerung ist jeweils mindestens eine mikrobiologische Kontrolluntersuchung des Kulturmediums der Spenderhornhaut durchzuführen. Die Eignung der gewählten Methode für das Untersuchungsmaterial ist zu belegen (mit Verweis auf die Kapitel 2.6.1 und 2.6.27 der Europäischen Pharmakopöe, Method Suitability). Fachlich begründete Abweichungen in den Testverfahren sind im Genehmigungsantrag darzulegen. Das Probenvolumen ist so zu wählen, dass eine größtmögliche Sensitivität des Testverfahrens gewährleistet ist.
Die Entnahme der ersten Mediumprobe für die mikrobiologische Untersuchung sollte für die Organkultur am Tag 5 ± 2 der Kultivierung erfolgen. Bei kurzzeitkultivierten Hornhäuten kann der mikrobiologische Kontrolltest vorgezogen werden.
Aufgrund der begrenzten Lagerfähigkeit der Spenderhornhäute kann abweichend von den in den Kapiteln 2.6.1 bzw. 2.6.27 der Europäischen Pharmakopöe angegebenen Zeiten für die Dauer der Inkubation die Beurteilung nach dem „Negative-to-date“-Konzept erfolgen. Hierbei wird ein Zwischenergebnis eines noch nicht abgeschlossenen Kontrolltests für die Freigabe der Gewebezubereitung herangezogen. Die Inkubationsdauer soll bei Anwendung dieses Konzepts sowohl für Proben von in Organkultur aufbewahrten Spenderhornhäuten als auch bei der hypothermen Lagerung mindestens 48 Stunden betragen, bis eine freigaberelevante Beurteilung des Keimwachstums in den mikrobiologischen Kulturmedien entsprechend der angewandten Methode vorgenommen werden darf. Die Freigabe der Spenderhornhäute auf der Grundlage der Ergebnisse des mikrobiologischen Kontrolltests am Kulturmedium erfolgt somit bei organkultivierten Hornhäuten frühestens am Tag 5 der Kultivierung, bei kurzzeitkultivierten Hornhäuten gegebenenfalls zu einem früheren Zeitpunkt. Der mikrobiologische Test wird anschließend bis zum Tag 14, bzw. bei Verwendung eines Kulturautomatensystems mindestens bis zum 7. Tag, weitergeführt und täglich auf ein mögliches Keimwachstum hin bewertet.
Nur Spenderhornhäute, die in dem beschriebenen Test einen negativen mikrobiologischen Befund aufweisen, dürfen für die Verwendung am Menschen freigegeben werden.
Spenderhornhäute, bei denen die mikrobiologische Untersuchung des Kulturmediums einen Keimnachweis erbracht hat, werden unverzüglich aus dem Bestand genommen. An der mikrobiologisch auffälligen Probe erfolgen eine Speziesidentifizierung und eine Resistenzprüfung.
Bei Anwendung des „Negative-to-date“-Konzepts ist bei einem nachfolgenden, nach der Freigabe/Transplantation und während der weiteren Inkubation auftretenden Nachweis von Keimwachstum umgehend die transplantierende Einrichtung zu informieren, um eine kurzfristige Kontrolluntersuchung und gegebenenfalls Behandlung des Empfängers zu ermöglichen. Weiterhin ist eine Speziesidentifizierung durchzuführen sowie ein Resistogramm anzufertigen. Diese Ergebnisse sind umgehend an die transplantierende Einrichtung weiterzuleiten. In gleicher Weise ist bei positiven mikrobiologischen Befunden zu verfahren, die aus weiteren Proben des Kulturmediums resultieren.
Die Ergebnisse weiterführender mikrobiologischer Untersuchungen dienen neben den für eine eventuelle Therapie notwendigen Informationen auch der Rückverfolgung der Eintragswege von Mikroorganismen sowie der Aufdeckung systematischer Fehler und sind als Maßnahme zur Risikoeinschätzung bestimmter Verfahrensschritte und damit auch zur Vermeidung künftiger Kontaminationen anzusehen. Der mikrobiologische Befund wird zusammen mit dem Herstellungsprotokoll der Spenderhornhaut aufbewahrt.
Sollte sich die Partnerhornhaut einer kontaminierten Probe im Bestand befinden, so wird diese in Quarantäne belassen und einer wiederholten Testung auf Sterilität unterzogen. Im Falle eines unauffälligen Ergebnisses der wiederholten Testung kann diese Hornhaut zur Transplantation durch die verantwortliche Person nach § 20c AMG im Sinne einer begründeten Einzelfallentscheidung freigegeben werden.
Eine weitere mikrobiologische Testung so spät wie möglich während der Kultivierung und unter Anwendung des oben beschriebenen „Negative-to-date“-Konzepts wird empfohlen.
10 Vermeidung von Kontaminationen
Neben den mit den genannten Verfahren nachweisbaren Bakterien und Pilzen sind Kontaminationen von Augenhornhäuten auch durch mit den herkömmlichen Methoden nicht anzüchtbare Mikroorganismen einschließlich Mykoplasmen, Parasiten und Viren möglich.
Um das Risiko der Verunreinigung der Augenhornhäute durch Infektionserreger und als apathogen geltende Keime so gering wie möglich zu halten, sind entsprechende Maßnahmen bei Spenderauswahl, Spendertestung, Entnahme, Zwischenlagerung, Transport, Verarbeitung, Kultivierung und Abgabe zu ergreifen. Hierbei ist auch das Risiko einer Kontaminierung der Gewebe durch Personal, Umgebung (einschließlich Oberflächen der Entnahme- und Verarbeitungsbereiche), verwendete Behältnisse und Materialien sowie einer Kreuzkontamination zwischen einzelnen Spendern zu berücksichtigen.
Beim Umgang mit dem Gewebe bei der Entnahme und in der Gewebebank sind die Grundsätze des aseptischen Arbeitens strikt einzuhalten. Neben der Schaffung der räumlichen und technischen Voraussetzungen sind eine Analyse der hygienisch relevanten Arbeitsschritte, die Erstellung eines Hygiene- und Reinigungsplans sowie die Schulung der Mitarbeiter notwendig.
Das Gewebe darf nur mit sterilen Instrumenten und Materialien in direkten Kontakt kommen. Die Hände und andere Körperbereiche des Personals sind wirksam als Kontaminationsquelle auszuschalten, wobei sowohl Desinfektion und Bekleidung als auch die Handhabung der Instrumente und Gewebe eine wichtige Rolle spielen. Die Arbeitsschritte und der Materialgebrauch sind so zu gestalten, dass Kreuzkontaminationen vermieden werden.
11 Freigabe der Spenderhornhäute für die Anwendung am Menschen
Es muss sichergestellt sein, dass die Spenderhornhäute durch die verantwortliche Person oder deren Vertreter gemäß § 20c AMG nicht freigegeben werden, bevor die Anforderungen des § 38 AMWHV erfüllt sind. Dabei gelten folgende Spezifikationen:
- 1.
-
Vollständige Kontraindikationsprüfung des Spenders mit unauffälligem Ergebnis.
- 2.
-
Spendereinwilligung liegt vor.
- 3.
-
Postmortale Zeit ≤ 72 Stunden (bzw. ≤ 16 Stunden für Spenderhornhäute, die kurzzeitkultiviert werden) bis zum Beginn der Kultivierung.
- 4.
-
Negative serologische bzw. molekularbiologische Diagnostik des Spenders (Hepatitis B, Hepatitis C, HIV, Syphilis).
- 5.
-
Negativer mikrobiologischer Befund des Kulturmediums.
- 6.
-
Keine relevanten, mikroskopisch sichtbaren, pathologischen Veränderungen (relevante Veränderungen sind: Stromatrübungen, wenn diese zentral liegen und damit optisch relevant sind, Stromaverdünnungen, wenn diese im Transplantatbereich liegen und nicht sicher traumatischer Genese sind, Stromaveränderungen infektiöser Genese, Ablösung der Descemetmembran).
- 7.
-
Ausreichende Endothelzellvitalität und -morphologie (Abweichungen vom Normbereich sind: großflächige, zentrale Mehrzellnekrosen und deutliche zusammenhängende Endothelzellnekrosen, Einzelzellnekrosen bzw. faltenassoziierte gruppierte Endothelzellnekrosen, die mehr als 10 % der gesamten Endothelzellfläche betreffen, ausgeprägter Polymegatismus, ausgeprägter Pleomorphismus, weniger als die Hälfte mit endothelzelltypischer Morphologie, ausgeprägte Granulierung/Vakuolisierung, Guttae der Endothelzellschicht bzw. Cornea guttata, Endothelzellverlust während der Kultivierung von mehr als 25 %; ist die Endothelzelldichte zu Beginn der Kultivierung nicht zuverlässig bestimmbar und somit ein Verlust im Verlauf nicht sicher quantifizierbar, ist besonders auf das Maß an Polymegatismus und Pleomorphismus der Endothelzellen zu achten, das hinweisend auf einen erhöhten Endothelzellverlust sein kann).
- 8.
-
Definierte Endothelzellquantität (siehe unten).
- 9.
-
Kennzeichnung (siehe Nummer 12.2).
- 10.
-
Gesamtdauer der Kultur inklusive Entquellung darf 34 Tage bei organkultivierten und 14 Tage bei kurzzeitkultivierten (hypotherme Lagerung) Spenderhornhäuten nicht überschreiten.
Bis zur Freigabe befindet sich die Spenderhornhaut in Quarantäne.
Spenderhornhäute, bei deren Endprüfung eine zentrale Endothelzelldichte von mindestens 2 000 Endothelzellen pro mm2 ermittelt wurde, dürfen für alle Hornhauttransplantationen verwendet werden: perforierende Keratoplastik, perforierende Limbokeratoplastik, hintere lamelläre Keratoplastik, vordere lamelläre Keratoplastik, tektonische Keratoplastik und für Stroma-Patch. Das Behältnis ist entsprechend der Bezeichnung der genehmigten Gewebezubereitung (z. B. „Humane Augenhornhaut, organkultiviert“) zu kennzeichnen (siehe Nummer 12.2).
Spenderhornhäute, bei deren Endprüfung eine Endothelzelldichte zwischen 1 000 und 2 000 Endothelzellen pro mm2 ermittelt wurde, dürfen nur für vordere lamelläre Keratoplastik, tektonische Keratoplastik, Stroma-Patch und perforierende Keratoplastik für temporären Hornhautersatz verwendet werden. Für diese Spenderhornhäute mit einer Endothelzelldichte unter 2 000 Zellen pro mm2 ist eine eigenständige Genehmigung nach § 21a AMG erforderlich. Das Behältnis ist entsprechend der Bezeichnung der genehmigten Gewebezubereitung (z. B. „Temporärer/stromaler Hornhautersatz, organkultiviert“) zu kennzeichnen (siehe Nummer 12.2). Augenhornhäute, die Trübungen oder Verdünnungen im Stroma nicht-infektiöser Genese aufweisen, können für endotheliale Transplantationen verwendet werden.
Es müssen gemäß § 38 Absatz 1 AMWHV Standardarbeitsanweisungen (Prüfanweisungen) vorliegen, welche die Kriterien, Verantwortlichkeiten und Verfahren für die Freigabe von Spenderhornhäuten genau angeben. Die endgültige Freigabe zur Transplantation muss gemäß § 38 Absatz 1 AMWHV durch die verantwortliche Person gemäß § 20c AMG durchgeführt und unterzeichnet werden. Das Verfahren muss die versehentliche Freigabe von Gewebe und Gewebezubereitungen verhindern, wenn die Voraussetzungen nach § 38 Absatz 2 AMWHV nicht erfüllt sind.
Die Aufzeichnungen müssen vor Freigabe der Spenderhornhäute nachweisen, dass alle einschlägigen Spezifikationen erfüllt wurden, dass insbesondere alle aktuellen Meldevordrucke, einschlägige medizinische Aufzeichnungen, Verarbeitungsaufzeichnungen und Testergebnisse sowie die Endverpackung überprüft worden sind. Bei gemäß § 21a AMG genehmigten Gewebezubereitungen beinhaltet die Freigabe auch die Bestätigung, dass die Gewebezubereitungen den Genehmigungsunterlagen entsprechend gewonnen, verarbeitet und geprüft wurden und alle freigaberelevanten Anforderungen erfüllt sind.
Die Hornhautbank definiert und implementiert Verfahren für die Kontrolle der Verpackungs- und Lagerungsbereiche, um schädliche Einflüsse auf die Funktionalität und Sicherheit der Gewebe und Gewebezubereitungen zu vermeiden.
Grundsätzlich sollte die Partnerhornhaut eines Hornhauttransplantats, bei dem eine schwerwiegende Reaktion eingetreten ist bzw. das möglicherweise zu einem schwerwiegenden Zwischenfall beim Empfänger geführt hat, nicht für die Anwendung am Menschen freigegeben werden (siehe Nummer 13, Dokumentations- und Meldepflichten bei schwerwiegenden unerwünschten Reaktionen und schwerwiegenden Zwischenfällen sowie Rückverfolgung und Rückruf).
12 Transport (inklusive Kennzeichnung) und Anwendung
Für den Transport von Gewebe gilt § 20c AMG in Verbindung mit § 39 AMWVH. Für den Transport in geeignetem Entquellungs-/Transportmedium darf die maximale Höchstdauer von sechs Tagen nicht überschritten werden; die nachfolgend aufgeführten Temperaturen müssen eingehalten werden. Grundsätzlich sind bei den organkultivierten Spenderhornhäuten Lagerungstemperaturen während des Transportes unter 10 °C und über 40 °C und für kurzzeitkultiverte Spenderhornhäute unter 2 °C und über 8 °C zu vermeiden, da diese zu einer Schädigung des Hornhautgewebes führen können und damit die Sicherheit für den Empfänger gefährden können.
Das Behältnis/die Packung muss gewährleisten, dass die Spenderhornhaut in dem spezifizierten Zustand steril erhalten bleibt. Die Zweckmäßigkeit aller Behältnisse und Packungen ist zu validieren bzw. es sind entsprechend validierte Behältnisse zu verwenden. Der Behälter muss so verschlossen werden, dass eindeutig erkennbar ist, wenn er geöffnet wurde (z. B. durch das Anbringen von Siegelaufklebern). Die Verpackung muss stabil und sicher sein.
Die Packung und die Art des Transportes müssen so gewählt werden, dass die Einhaltung einer Temperatur des Transportmediums sicher gewährleistet ist. Dies ist durch die Hornhautbank in regelmäßigen Abständen zu überprüfen. Sollte eine Temperaturstabilität durch die Packung oder den Transportweg auch in möglichen Fällen unerwartet hoher oder sehr niedriger Außentemperaturen nicht zweifelsfrei gesichert werden können (Transportvalidierung), so ist ein Temperaturmessgerät beizulegen, welches mindestens halbstündlich die Temperatur im Inneren der Packung misst und die Daten speichert. Das Temperaturmessgerät sollte dabei so gewählt werden, dass bei der Entgegennahme der Sendung am Bestimmungsort unproblematisch eine Über- bzw. Unterschreitung der zulässigen Temperatur des Transportmediums erkannt werden kann. Die Hornhautbank hat die Personen am Bestimmungsort des Hornhauttransplantats über die Bedienung des Temperaturmessgerätes zu informieren (z. B. durch Beilegen einer Anleitung). Sollte das festgelegte Temperaturintervall nicht eingehalten worden sein, so hat der Operateur bzw. eine durch diesen beauftragte Person, die Hornhautbank vor der geplanten Transplantation zu kontaktieren. Der Anwender/Operateur hat auf Basis einer Nutzen-Risiko-Bewertung für den Patienten zu entscheiden, ob die Spenderhornhaut trotz Nichteinhaltung der vorgeschriebenen Transporttemperatur transplantiert werden kann. Jede Hornhautbank stellt sicher, dass ein genaues, zügiges und überprüfbares Verfahren vorhanden ist, mit dem sie jedes Gewebe von der Verteilung zurückziehen kann, das mit einem schwerwiegenden Zwischenfall oder einer schweren unerwünschten Reaktion in Verbindung stehen könnte. Dies umfasst auch die Meldung an die zuständige Behörde, das Paul-Ehrlich-Institut oder die zuständige Landesbehörde. Innerhalb der Hornhautbank ist die verantwortliche Person gemäß § 20c AMG dafür verantwortlich, alle bekannt gewordenen Meldungen über schwerwiegende unerwünschte Reaktionen zu sammeln, die Notwendigkeit eines Rückrufs (siehe Nummer 13) zu bewerten und die nötigen Maßnahmen zu koordinieren.
Die Hornhautbank ist für den ordnungsgemäßen Transport verantwortlich. Sollte für den Transport die Hilfe Dritter in Anspruch genommen werden, so muss eine vertragliche Vereinbarung die Verantwortlichkeiten regeln.
Der Spenderhornhaut ist ein Rückmeldebogen (Transplantationsbegleitschein) für den klinischen Anwender des Hornhauttransplantats beizulegen, der nach Abschluss der Behandlung, spätestens sechs Monate postoperativ, an die Hornhautbank zurückzusenden ist. Dieser muss die notwendigen Informationen für die Rückverfolgbarkeit und sollte Daten zur Erfassung und Auswertung der klinischen Resultate beinhalten.
12.1 Abgabe und Anwendung
Die Hornhauttransplantate werden als Ersatz für die aufgrund einer Erkrankung getrübte, verformte oder andersartig irreversibel funktionsgestörte Hornhaut eingesetzt. Die Indikation zur Transplantation einer Augenhornhaut stellt der Operateur. Dieser wählt auch das operative Verfahren (beispielsweise perforierende oder lamelläre Transplantation) aus.
Hinsichtlich der regulatorischen Anforderungen an das Verfahren der lamellären Keratoplastik wird grundsätzlich unterschieden zwischen einer prospektiven Herstellung in der Hornhautbank und einer Herstellung während der Operation.
Sofern bereits lamelläre Hornhauttransplantate wie z. B. Descemet Membrane Endothelial Keratoplasty (DMEK)-Transplantate von der Augenhornhautbank zur Verfügung gestellt werden, sind diese Bearbeitungsschritte inklusive der verwendeten Geräte wie z. B. eines Mikrokeratoms Teil des Be- und Verarbeitungsverfahrens und fallen unter die Voraussetzungen für die Antragstellung bei der zuständigen Landesbehörde bzw. beim Paul-Ehrlich-Institut.
Im Gegensatz dazu ist nach dem Inverkehrbringen der Augenhornhaut der Einsatz technischer Geräte wie des Mikrokeratoms im Rahmen der Anwendung an der Augenhornhaut unter der Verantwortung des anwendenden Arztes für die Transplantation nicht genehmigungspflichtig.
Für die Allokation von Spenderhornhäuten wurden keine gesetzlichen Regelungen getroffen. Die regionale Abgabe nach folgenden Kriterien (Prioritätenliste) wird empfohlen (dabei ist zu beachten, dass eine Genehmigung gemäß § 21a AMG vorliegen muss, falls eine Gewebezubereitung in den Verkehr gebracht wird):
- 1.
-
Notfallsituationen innerhalb, aber auch außerhalb des Krankenhauses, dem die Hornhautbank angeschlossen ist, sollen höchste Priorität haben (z. B. perforierte Hornhautulzera oder schmerzhafte Augenerkrankungen).
- 2.
-
Kinder und Jugendliche sollen vorrangig versorgt werden.
- 3.
-
Patienten mit nur einem potenziell funktionierenden Auge sollen vorrangig versorgt werden.
- 4.
-
Unter den verbliebenen Patienten sollen die Erfolgsaussichten und die Wartezeit berücksichtigt werden.
Transplantate, die nach diesen Kriterien nicht vergeben werden, können über einen geeigneten Weg abgegeben werden. Auf die Möglichkeit der Nachfrage bei Gewebenetzwerken innerhalb bzw. außerhalb der EU wird hingewiesen. Beim Verbringen und Import von Transplantaten sind bei der Einfuhr aus der EU bzw. Drittländern die erforderlichen gesetzlichen Vorgaben (u. a. § 21a Absatz 9, § 72b, § 73 Absatz 3a, § 39 Absatz 2 und 3, § 31a AMG und Abschnitt 5a und b AMWHV) zu beachten.
Es sollten Verfahren für den Umgang mit Anfragen nach Spenderhornhäuten vorhanden sein. Die Regeln für die Zuweisung von Spenderhornhäuten an bestimmte Personen oder Einrichtungen des Gesundheitswesens sind zu dokumentieren und den Betroffenen auf Anfrage zur Verfügung zu stellen.
Bei der Abgabe von Hornhauttransplantaten sollte zwischen dem klinischen Anwender/Operateur und der Hornhautbank eine schriftliche Vereinbarung getroffen werden. Hier sollten die Leistungen und Pflichten beider Parteien klar vereinbart werden.
Es muss ein Dokumentationssystem für den Umgang mit zurückgeschickten Spenderhornhäuten vorhanden sein.
12.2 Kennzeichnung für das Inverkehrbringen
Das Behältnis für Spenderhornhäute muss zum Inverkehrbringen Angaben gemäß § 10 Absatz 8b AMG in Verbindung mit § 36 Absatz 8 AMWHV (siehe Nummer 17.1 der Bezugsdokumente) tragen. Gewebe und Gewebezubereitungen, die zum Zweck der Anwendung beim Menschen in den Verkehr gebracht werden, müssen gemäß § 10 Absatz 8b AMG und Abschnitt 5 AMWHV auf dem Etikett des Primärbehältnisses mit einem Einheitlichen Europäischen Code (SEC) gekennzeichnet sein. Zusätzlich wird der SEC in den Begleitdokumenten gemäß § 41b Absatz 5 Satz 4 AMWHV vermerkt.
Kann eine der Informationen nicht auf dem Primärpackmittel angegeben werden, so ist sie auf einem gesonderten Blatt anzugeben, das so beizufügen ist, dass die eindeutige Zuordnung erhalten bleibt.
Auf der äußeren Umhüllung – soweit verwendet – sind Informationen gemäß § 10 Absatz 8b AMG (siehe Nummer 17.1 der Bezugsdokumente) entweder auf dem Etikett für die äußere Umhüllung oder in beiliegenden Unterlagen (z. B. der Gebrauchs- und Fachinformation) anzugeben.
12.3 Kennzeichnung des Transportbehälters
Werden die Gewebe von einem Dritten transportiert, muss jeder Transportbehälter mindestens Angaben gemäß § 39 Absatz 5 AMWHV (siehe Nummer 17.1 der Bezugsdokumente) tragen.
13 Dokumentations- und Meldepflichten bei schwerwiegenden unerwünschten Reaktionen und schwerwiegenden Zwischenfällen sowie Rückverfolgung und Rückruf
Gemäß § 13a TPG haben die Einrichtungen der medizinischen Versorgung dafür zu sorgen, dass für Zwecke der Rückverfolgung oder für Zwecke der Risikoerfassung jedes übertragene Gewebe von dem behandelnden Arzt oder unter dessen Verantwortung nach Maßgabe des § 7 TPG-GewV dokumentiert wird.
Gemäß § 13b TPG haben die Einrichtungen der medizinischen Versorgung jeden schwerwiegenden Zwischenfall im Sinne des § 63i Absatz 6 AMG und jede schwerwiegende unerwünschte Reaktion im Sinne des § 63i Absatz 7 AMG, die bei oder nach der Übertragung der Gewebe beobachtet wurde und mit der Qualität und Sicherheit der Gewebe im Zusammenhang stehen kann, unverzüglich nach deren Feststellung zu dokumentieren und der Gewebeeinrichtung, von der sie das Gewebe erhalten haben, unverzüglich mit allen Angaben, die für die Rückverfolgung und für die Qualitäts- und Sicherheitskontrolle erforderlich sind, nach Maßgabe der §§ 8 und 9 TPG-GewV zu melden.
Nach § 63i Absatz 1 AMG hat der Inhaber einer Genehmigung für Gewebezubereitungen im Sinne von § 21a AMG Unterlagen über Verdachtsfälle von schwerwiegenden Zwischenfällen oder schwerwiegenden unerwarteten Reaktionen, die in den Mitgliedstaaten der Europäischen Union oder in den Vertragsstaaten des Abkommens über den Europäischen Wirtschaftsraum oder in einem Drittland aufgetreten sind, sowie über die Anzahl der Rückrufe zu führen. Auf dieser Grundlage hat der Inhaber der Genehmigung der zuständigen Bundesoberbehörde (Paul-Ehrlich-Institut) einen aktualisierten Bericht über die Unbedenklichkeit der Arzneimittel unverzüglich nach Aufforderung oder, soweit Rückrufe oder Fälle oder Verdachtsfälle schwerwiegender Zwischenfälle oder schwerwiegende unerwünschte Reaktionen betroffen sind, mindestens einmal jährlich vorzulegen.
Die Begriffe „schwerwiegender Zwischenfall“ und „schwerwiegende unerwünschte Reaktion“ sind in § 63i Absatz 6 bzw. Absatz 7 AMG legal definiert.
Gemäß § 63i Absatz 2 AMG hat der Inhaber einer Zulassung oder Genehmigung für Gewebezubereitungen jeden Verdacht eines schwerwiegenden Zwischenfalls und jeden Verdacht einer schwerwiegenden unerwünschten Reaktion zu dokumentieren und unverzüglich, spätestens innerhalb von 15 Tagen nach Bekanntwerden, der zuständigen Bundesoberbehörde anzuzeigen. Welche Angaben diese Anzeige enthalten muss, regelt § 63i Absatz 2 Satz 2 und 3 AMG. Die Meldung hat an das Paul-Ehrlich-Institut zu erfolgen. Meldeformulare sind über den Internetauftritt des Paul-Ehrlich-Instituts abrufbar.
Im Übrigen sind die Gewebeeinrichtungen nach § 63i Absatz 3 Satz 1 AMG für alle nicht nach § 21 AMG bzw. § 21a AMG zulassungs- bzw. genehmigungspflichtigen Augenhornhäute sowie Gewebe zur Meldung an die jeweils zuständige Landesbehörde verpflichtet. Die notwendigen Angaben bestimmt § 63i Absatz 3 Satz 2 und 3 AMG.
Daneben sind gemäß § 40 Absatz 1 AMWHV die betroffenen Gewebeeinrichtungen nach vorher erstellter Standardarbeitsanweisung von den Entnahmeeinrichtungen nach § 20b AMG über alle bekannt gewordenen schwerwiegenden unerwünschten Reaktionen im Sinne von § 63i Absatz 7 AMG und entsprechende Verdachtsfälle, die die Qualität und Sicherheit der Gewebe oder Gewebezubereitungen beeinflussen oder auf diese zurückgeführt werden können, unverzüglich in Kenntnis zu setzen.
Entsprechendes gilt gemäß § 40 Absatz 2 AMWHV für Entnahmeeinrichtungen und Gewebespenderlabore im Falle von schwerwiegenden Zwischenfällen im Sinne von § 63i Absatz 6 AMG und entsprechenden Verdachtsfällen, die im Zusammenhang mit der Gewinnung oder den für die Gewinnung erforderlichen Laboruntersuchungen aufgetreten sind. In Gewebeeinrichtungen ist die verantwortliche Person nach § 20c AMG dafür verantwortlich, dass alle bekannt gewordenen Meldungen über schwerwiegende unerwünschte Reaktionen und schwerwiegende Zwischenfälle oder Verdachtsfälle solcher Reaktionen oder Zwischenfälle entsprechend gesammelt und bewertet werden und den zuständigen Behörden (vgl. § 63i Absatz 2 und 3 AMG) unverzüglich übermittelt werden. Der Inhalt der Meldungen ergibt sich aus § 40 Absatz 3 und 4 AMWHV.
Die verantwortliche Person nach § 20c AMG hat gemäß § 40 Absatz 5 AMWHV auch dafür Sorge zu tragen, dass Gewebe und Gewebezubereitungen entsprechend identifiziert, ausgesondert und zurückgerufen werden können. Sie hat nach schriftlich festgelegten Verfahren die Notwendigkeit eines Rückrufs zu bewerten und die nötigen Maßnahmen innerhalb vorher festgelegter Zeiträume zu koordinieren sowie die zuständige Behörde über jeden Rückruf unverzüglich zu unterrichten und dabei auch mitzuteilen, an welche Einrichtungen die Gewebe oder Gewebezubereitungen ausgeliefert wurden und welche Maßnahmen sie in Bezug auf andere möglicherweise betroffene Gewebe ergriffen hat. Die Wirksamkeit der Verfahren ist regelmäßig zu überprüfen, beispielsweise im Rahmen eines internen Qualitätsmanagement-Audits. Für die Gewebeeinrichtung ist § 13c TPG zu beachten. Insbesondere hat eine Gewebeeinrichtung oder eine Einrichtung der medizinischen Versorgung bei begründetem Verdacht, dass Gewebe eine schwerwiegende Krankheit auslösen kann, der Ursache unverzüglich nachzugehen und das Gewebe von dem Spender zu dem Empfänger oder umgekehrt zurückzuverfolgen. Sie hat ferner vorangegangene Gewebespenden des Spenders zu ermitteln, zu untersuchen und zu sperren, wenn sich der Verdacht bestätigt.
Grundsätzlich sollte die Partnerhornhaut eines Hornhauttransplantats, bei dem eine schwerwiegende Reaktion beim Empfänger eingetreten ist, nicht für die Anwendung am Menschen freigegeben werden.
Wird in einer Hornhautbank ein schwerwiegender Zwischenfall oder eine schwerwiegende unerwünschte Reaktion erst nach Transplantation beider Spenderhornhäute bekannt, so sind die betroffenen transplantierenden Einrichtungen von der Hornhautbank unverzüglich mündlich und schriftlich darüber zu informieren, damit gegebenenfalls notwendige Maßnahmen zum Schutz des Empfängers (z. B. außerplanmäßige klinische Kontrolluntersuchung, Entfernung/Austausch des Spendergewebes) ohne Zeitverlust eingeleitet werden können.
Übersicht über die Dokumentations- und Meldepflichten der Einrichtungen der medizinischen Versorgung, in denen Spenderhornhäute übertragen werden, und der Hornhautbanken
| Ereignis | unverzüglich zu melden |
|---|---|
| schwerwiegende unerwünschte Reaktionen (§ 63i Absatz 7 AMG) und Verdachtsfälle solcher Reaktionen | durch Person nach § 20b Absatz 1 Satz 3 Nummer 1 AMG an betroffene (belieferte) Gewebeeinrichtung
(§ 40 Absatz 1 AMWHV) bei genehmigungspflichtigen Gewebezubereitungen durch verantwortliche Person nach § 20c AMG spätestens innerhalb von 15 Tagen an PEI als der zuständigen Bundesoberbehörde (§ 63i Absatz 2 AMG, § 40 Absatz 3 AMWHV) bei nicht genehmigungspflichtigen Gewebezubereitungen durch verantwortliche Person nach § 20c AMG an zuständige Behörde (§ 63i Absatz 3 AMG, § 40 Absatz 3 AMWHV) |
| schwerwiegende Zwischenfälle (§ 63i Absatz 6 AMG) und Verdachtsfälle schwerwiegender Zwischenfälle | durch Person nach § 20b Absatz 1 Satz 3 Nummer 1 AMG an betroffene (belieferte) Gewebeeinrichtung
(§ 40 Absatz 2 AMWHV) bei genehmigungspflichtigen Gewebezubereitungen durch verantwortliche Person nach § 20c AMG spätestens innerhalb von 15 Tagen an PEI als der zuständigen Bundesoberbehörde (§ 63i Absatz 2 AMG, § 40 Absatz 4 AMWHV) bei nicht genehmigungspflichtigen Gewebezubereitungen durch verantwortliche Person nach § 20c AMG an zuständige Behörde (§ 63i Absatz 3 AMG, § 40 Absatz 4 AMWHV) |
14 Verbot des Handels mit Spenderaugen und Spenderhornhäuten
Gemäß § 17 Absatz 1 Satz 1 TPG ist der Handel mit Organen und Geweben wie menschlichen Spenderaugen bzw. Spenderhornhäuten verboten. Die Gewährung oder Annahme eines angemessenen Entgelts für die zur Erreichung des Ziels der Heilbehandlung gebotenen Maßnahmen, insbesondere für die Entnahme, die Konservierung, die weitere Aufbereitung einschließlich der Maßnahmen zum Infektionsschutz, die Aufbewahrung und die Beförderung der Gewebe ist gemäß § 17 Absatz 1 Satz 2 Nummer 1 TPG zulässig.
Gemäß § 17 Absatz 2 TPG ist es ebenso verboten, Gewebe, für die ein Handelsverbot nach § 17 Absatz 1 Satz 1 TPG besteht, zu entnehmen, auf einen anderen Menschen zu übertragen oder sich übertragen zu lassen. Hinsichtlich der weiteren Straf- und Bußgeldvorschriften wird auf die §§ 18 bis 20 TPG verwiesen.
15 Import von Spenderaugen und Spenderhornhäuten aus dem Ausland
Wird ein Transplantat in den Verkehr gebracht, muss eine Genehmigung nach § 21a Absatz 1 AMG oder beim Verbringen aus einem EU-Land eine Bescheinigung gemäß § 21a Absatz 9 AMG des Paul-Ehrlich-Instituts als zuständiger Bundesoberhörde gemäß § 77 Absatz 2 AMG vorliegen.
Wenn eine Hornhautbank Gewebe gemäß § 1a Nummer 4 TPG oder Gewebezubereitungen gemäß § 4 Absatz 30 AMG zum Zwecke der Abgabe an andere oder zur Be- und Verarbeitung aus Drittstaaten einführt, müssen u. a. die Anforderungen gemäß den §§ 72b, 73 Absatz 3a AMG erfüllt sein (siehe Nummer 17.1 der Bezugsdokumente).
16 Anlagen
16.1 Spenderausschlusskriterien (Anlage 1)
Die Auswahlkriterien für Spender beruhen auf einer Risikoanalyse im Zusammenhang mit der Verwendung der Spenderaugen bzw. -hornhäute. Anzeichen für solche Risiken sind durch Anamnese, zweckdienliche Quellen wie Krankenakten und Befragung behandelnder Ärzte, biologische Untersuchung, postmortale Untersuchung und sonstige geeignete Untersuchungen zu ermitteln. Sofern die Spende nicht aufgrund einer dokumentierten Risikobewertung, die von dem Arzt gemäß § 8d TPG durchgeführt wird, gerechtfertigt ist, sind Spender von der Spende auszuschließen, sofern es Hinweise auf eines der folgenden Ausschlusskriterien gibt, die in der gemäß § 3 Absatz 1 TPG-GewV für Augenhornhäute spezifizierten Anlage 1 der TPG-GewV aufgeführt sind:
- 1.
-
Postmortale Zeit über 72 Stunden für Spenderhornhäute, die organkultiviert werden.
- 2.
-
Postmortale Zeit über 16 Stunden für Spenderhornhäute, die kurzzeitkultiviert (hypotherme Lagerung) werden.
- 3.
-
Unbekannte Todesursache, sofern die Todesursache nicht nach der Entnahme aus der Autopsie hervorgeht und kein anderes in diesem Abschnitt genanntes allgemeines Ausschlusskriterium zutrifft.
- 4.
-
Erkrankung unbekannter Ätiologie in der Vorgeschichte.
- 5.
-
Zentralnervöse Erkrankungen unklarer Genese, wie z. B.:
- a)
-
Multiple Sklerose.
- b)
-
Amyotrophe Lateralsklerose.
- c)
-
Morbus Alzheimer.
- d)
-
Retrovirale ZNS-Erkrankung.
- e)
-
Morbus Parkinson.
- 6.
-
Spender mit malignen Erkrankungen können für Bulbus- und Hornhautspenden in Betracht kommen, ausgenommen Spender mit Retinoblastom, hämatologischen Neoplasien und malignen Tumoren des Augenhintergrunds.
- 7.
-
Risiko der Krankheitsübertragung durch Prionen. Dieses Risiko besteht bei:
- a)
-
Personen, bei denen die Creutzfeldt-Jakob-Krankheit oder die neue Variante der Creutzfeldt-Jakob-Krankheit diagnostiziert wurde, oder die eine nicht iatrogene Creutzfeldt-Jakob-Krankheit in der familiären Vorgeschichte aufweisen. Bei der neuen Variante der Creutzfeldt-Jakob-Krankheit sind eventuell weitere Vorsichtsmaßnahmen zu empfehlen, beispielsweise die Berücksichtigung eines kumulativen Aufenthalts in Großbritannien von mehr als sechs Monaten zwischen 1980 und 1996.
- b)
-
Personen mit anamnestisch erhobener rasch fortschreitender Demenz oder einer degenerativen neurologischen Erkrankung unbekannter Ursache.
- c)
-
Empfänger von Hormonen, die aus der menschlichen Hypophyse gewonnen wurden (z. B. Wachstumshormone).
- d)
-
Empfänger von Transplantaten von Cornea, Sklera und Dura mater.
- e)
-
Personen, die nicht dokumentierten neurochirurgischen Operationen unterzogen wurden, bei denen möglicherweise Dura mater verwendet wurde.
- 8.
-
Systemische Infektionen, die zum Zeitpunkt der Spende nicht unter Kontrolle sind, einschließlich bakterieller Infektionen, systemischer viraler, Pilz- oder parasitärer Infektionen, oder signifikante lokale Infektionen in den zu spendenden Geweben;Spender mit bakterieller Sepsis können für eine Augenhornhautspende beurteilt und in Betracht gezogen werden, allerdings nur, sofern die Hornhäute in einer Organkultur aufbewahrt werden, welche den Nachweis einer etwaigen bakteriellen Kontamination des Gewebes ermöglicht.
- 9.
-
Ausschluss von Personen, bei denen jemals folgende Infektionen nachgewiesen wurden:
- a)
-
Viral: Anamnestisch erhobene, klinisch oder durch bestätigte Labortests nachgewiesene HIV-Infektion, Übertragungsrisiko akuter oder chronischer Hepatitis B (außer bei Personen mit nachgewiesenem Immunstatus), Hepatitis C und HTLV I/II oder Anzeichen von Risikofaktoren für diese Infektionen.
- b)
-
Protozoonosen: Babesiose, Trypanosomiasis (z. B. Chagas-Krankheit), Leishmaniose.
- c)
-
Syphilis und andere chronisch persistierende bakterielle Infektionen, wie Brucellose, Fleckfieber und andere Rickettsiosen, Lepra, Rückfallfieber, Melioidose oder Tularämie.
- 10.
-
Ausschluss bei Personen mit Hinweisen auf folgende Erkrankungen:
- a)
-
Für zwei Jahre nach Infektion mit Salmonella typhi und S. paratyphi bzw. nach dokumentierter Heilung von Q-Fieber, Tuberkulose, Leptospirosen.
- b)
-
Für vier Jahre nach dokumentierter Heilung von Malaria.
- c)
-
Für mindestens vier Wochen nach Abklingen der Symptome von Masern, Röteln, Varicella-Zoster, Hepatitis und anderer als der oben erwähnten Infektionskrankheiten, viraler Meningitis, viraler Enzephalitis, viralem hämorrhagischem Fieber.
- 11.
-
Lokal: Signifikante lokale Infektion der Augen durch Bakterien, Viren, Parasiten oder Pilze.
- 12.
-
Anzeichen für ungültige Testergebnisse der Spenderblutproben, wegen:
- a)
-
Hämodilution (Beispiel in Anlage 2, siehe Nummer 16.2): Gewebeeinrichtungen dürfen Gewebe von Spendern mit mehr als 50 %iger Plasmaverdünnung (siehe Nummer 16, z. B. durch Infusion von Kristalloiden innerhalb einer Stunde vor Blutentnahme/Tod bzw. Gabe von Kolloiden oder Blutprodukten innerhalb von 48 Stunden vor Blutentnahme/Tod) nur annehmen, wenn die angewendeten Untersuchungsmethoden für solches Plasma validiert sind oder wenn eine Prätransfusionsprobe vorliegt.
- b)
-
Behandlung mit immunsuppressiven Wirkstoffen.
- 13.
-
Anzeichen sonstiger Risikofaktoren für Infektionskrankheiten auf der Grundlage einer Risikobewertung unter Berücksichtigung der Reisen und der Expositionsgeschichte des Spenders sowie der lokalen Prävalenz von Infektionskrankheiten.
- 14.
-
Personen, deren Sexualverhalten ein gegenüber der Allgemeinbevölkerung deutlich erhöhtes Übertragungsrisiko für durch Gewebe übertragbare schwere Infektionskrankheiten wie HBV, HCV oder HIV bergen.
- 15.
-
Anzeichen am Körper des Spenders, die ein Infektionsrisiko nahelegen.
- 16.
-
Aufnahme oder Exposition gegenüber einer Substanz (wie Zyanid, Blei, Quecksilber, Gold), die auf den Empfänger in einer gesundheitsschädlichen Dosis übertragen werden könnte.
- 17.
-
Kürzlich erfolgte Impfung mit einem Lebendimpfstoff aus abgeschwächtem Virus, bei der eine Übertragung für möglich gehalten wird (z. B. Tollwut).
- 18.
-
Empfänger von Heterotransplantaten und Xenotransplantaten.
16.2 Beispiel für Algorithmus zur Berechnung der Hämodilution (Anlage 2)
| Protokoll zur Ermittlung der Hämodilution |
|
(Gewebespende)
|
| Spendernummer: |
Entnehmende Einrichtung: |
|
|
|
|
- –
-
Der Gewebespender hat vor dem Tod relevante Infusionen/Transfusionen erhalten.
- –
-
Die für die Laboruntersuchungen entnommene Blutprobe wurde nach diesen Infusionen/Transfusionen gemäß den Vorgaben der TPG-GewV gewonnen.
- –
-
Daher ist die eingetretene Blut- bzw. Plasmaverdünnung zu ermitteln und festzustellen, ob die nach den Infusionen entnommene Blutprobe für die Screeninguntersuchungen geeignet ist.
- –
-
Dem Gewebespender wurden folgende Lösungen/Blutprodukte infundiert/transfundiert:Kristalloide Lösungen:
Zeitraum vor der Entnahme
der BlutprobeInfundiertes Volumen
in mlVerbleibendes Volumen
in %Verbleibendes Volumen
in ml> 24 h 0 2 – 24 h 25 1 – 2 h 50 < 1 h 75 Gesamtes verbleibendes Volumen in ml (V1): Blut, Blutpräparate, kolloidale Lösungen:Zeitraum vor der Entnahme
der BlutprobeInfundiertes Volumen
in mlVerbleibendes Volumen
in %Verbleibendes Volumen
in ml24 – 48 h
(Blut, Blutpräparate)100 24 – 48 h
(kolloidale Lösungen)50 0 – 24 h 100 Gesamtes verbleibendes Volumen in ml (V2): Berechnung der Prozentualen Blut- bzw. Plasmaverdünnung:Geschätztes Gesamt-Blutvolumen (GGB)
Geschätztes Plasmavolumen (PV)Prozentuale Blut- bzw. Plasmaverdünnung (PBV bzw. PPV) GBB = 70 ml x kg Körpergewicht
PV = 42 ml x kg Körpergewicht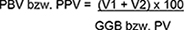
- –
-
Die Blutprobe ist für die Laboruntersuchungen geeignet, wenn die prozentuale Blut- bzw. Plasmaverdünnung ≤ 50 % ist.
- –
-
Die Blutprobe ist für die Laboruntersuchungen nicht geeignet, wenn die prozentuale Blut- bzw. Plasmaverdünnung > 50 % ist.
| Die Blutprobe ist geeignet: |
ja ☐ nein ☐ | |
|
|
|
|
| Unterschrift | Datum | |
17 Anhang
17.1 Bezugsdokumente
Transplantationsgesetz (TPG) in der jeweils geltenden Fassung;
http://www.gesetze-im-internet.de/tpg/
TPG-Gewebeverordnung (TPG-GewV) in der jeweils geltenden Fassung; http://www.gesetze-im-internet.de/tpg-gewv/BJNR051200008.html
Arzneimittelgesetz (insbesondere die §§ 1 bis 4, 4a, 13, 20b bis 20d, 21, 21a, 63, 64, 72b und 142 AMG) in der jeweils geltenden Fassung;
http://www.gesetze-im-internet.de/amg_1976/
Arzneimittel- und Wirkstoffherstellungsverordnung (insbesondere Nummer 1, 2, 5a und 5b AMWHV) in der jeweils geltenden Fassung;
http://www.gesetze-im-internet.de/amwhv/
Meldeformulare für SAE/SAR (http://www.pei.de/meldeformulare-human)
Berichtspflicht gemäß § 8d TPG; www.pei.de/tpg-8.de
17.2 Literaturverzeichnis
- 1.
-
Arbeitsrichtlinien (Gute Fachliche Praxis für Hornhautbanken) der Sektion Gewebetransplantation und Biotechnologie der Deutschen Ophthalmologischen Gesellschaft. Der Ophthalmologe 2009; 106:265 – 74
- 2.
-
Armitage WJ: Developments in Corneal Preservation in Essentials in Ophthalmology, Corneal and External Eye Disease. Springer Verlag 2008
- 3.
-
Bestattungsgesetze der Länder über http://www.aeternitas.de/inhalt/recht/ (16. November 2016)
- 4.
-
Borderie VM: Donor selection, retrieval and preparation of donor tissue. Donor selection. Dev Ophthalmol 2009; 43:22-30
- 5.
-
Bundesärztekammer – Wissenschaftlicher Beirat: Richtlinie gemäß § 16 Absatz 1 Satz 1 Nummer 1 TPG für die Regeln zur Feststellung des Todes nach § 3 Absatz 1 Satz 1 Nummer 2 TPG und die Verfahrensregeln zur Feststellung des endgültigen, nicht behebbaren Ausfalls der Gesamtfunktion des Großhirns, des Kleinhirns und des Hirnstamms nach § 3 Absatz 2 Nummer 2 TPG, Vierte Fortschreibung. Dtsch Arztebl 2015, DOI: 10.3238/arztebl.2015.rl_hirnfunktionsausfall_01
- 6.
-
Bundesärztekammer – Wissenschaftlicher Beirat: Richtlinien zum Führen einer Hornhautbank. Dtsch Arztebl 2000; 97(14):A 2122 – 24
- 7.
-
Bundesärztekammer – Wissenschaftlicher Beirat: Richtlinien zum Führen einer Knochenbank. Dtsch Arztebl 2001; 98(15):A 1011 – 16
- 8.
-
Chen JY, Jones MN, Srinivasan S, Neal TJ, Armitage WJ, Kaye SB: Endophthalmitis after penetrating keratoplasty. Ophthalmology 2015; 122(1):25 – 30
- 9.
-
Deutscher Ethikrat: Humanbiobanken für die Forschung, Stellungnahme. Berlin 2010; über http://www.ethikrat.org/
- 10.
-
Durchführungsbestimmungen der Arbeitsgemeinschaft Deutscher Hornhautbanken für die Kultivierung von Spender-Hornhäuten und die Organisation von Hornhautbanken vom 30. September 2001;
http://www.dog.org/wp-content/uploads/2009/09/durchfuehrungsbest_hornhautbank.pdf - 11.
-
Ehlers N, Hjortdal J, Nielsen K: Corneal grafting and banking. Dev Ophthalmol 2009; 43:1 – 14
- 12.
-
European Eye Bank Association (EEBA): Agreements on Minimum Standards. https://www.eeba.eu/portal
- 13.
-
European Pharmacopoeia 8.8: Microbiological control of cellular products. 2016; 2.6.27
- 14.
-
European Pharmacopoeia 8.8: Minimising the risk of transmitting animal spongiforme encephalopathy agents via human and veterinary medical products. 2016; 5.2.8
- 15.
-
European Pharmacopoeia 8.8: Sterility. 2016; 2.6.1
- 16.
-
Eye Bank Association of America (EBAA): Medical Standards. November 2007; http://restoresight.org/
- 17.
-
Fuest M, Plum W, Salla S, Walter P, Hermel M: Conjunctival and intraocular swabs for the microbiological assessment of donor corneas. Acta ophthalmologica 2016; 94(1):70 – 5
- 18.
-
Li S, Bischoff M, Schirra F, Langenbucher A, Ong M, Halfmann A et al.: Correlation between microbial growth in conjunctival swabs of corneal donors and contamination of organ culture media. Der Ophthalmologe 2014; 111(6):553 – 9
- 19.
-
Maier P, Reinhard T: Kapitel 4.3 Augenhornhäute. In: Pühler W, Middel C-D, Hübner M (Hrsg): Praxisleitfaden Gewebegesetz, Deutscher Ärzteverlag 2009
- 20.
-
Pels E, Beele H, Claerhout I: Eye bank issues: II. Preservation techniques: warm versus cold storage. Int Ophthalmol 2008; 28(3):155 – 63
- 21.
-
Pels E, Pollock G: Storage of Donor Cornea for Penetrating and Lamellar Transplantation, Corneal Disease, Recent Developments in Diagnosis and Therapy. Springer Verlag 2013
- 22.
-
Pels E, Rijneveld WJ: Organ culture preservation for corneal tissue. Technical and quality aspects. Dev Ophthalmol 2009; 43:31 – 46
- 23.
-
Richtlinie 2004/23/EG des Europäischen Parlamentes und des Rates vom 31. März 2004 zur „Festlegung von Qualitäts- und Sicherheitsstandards für die Spende, Beschaffung, Testung, Verarbeitung, Konservierung, Lagerung und Verteilung von menschlichen Geweben und Zellen“, zuletzt geändert durch Verordnung (EG) Nr. 596/2009 des Europäischen Parlamentes und des Rates vom 18. Juni 2009. ABl. L 188:14. Official Journal of the European Union, L102/58, 7. April 2005
- 24.
-
Richtlinie 2006/17/EG der Kommission der Europäischen Gemeinschaft vom 8. Februar 2006 zur Umsetzung der Richtlinie 2004/23/EG hinsichtlich technischer Vorschriften für die Spende, Beschaffung und Testung von menschlichen Geweben und Zellen, zuletzt geändert durch Richtlinie 2012/39/EU der Kommission vom 26. November 2012; ABl. L 327:24, Berichtigung ABl. L 98:11
- 25.
-
Richtlinie 2006/86/EG der Kommission der Europäischen Gemeinschaft vom 24. Oktober 2006 zur Umsetzung der Richtlinie 2004/23/EG hinsichtlich der Anforderung an die Rückverfolgbarkeit, der Meldung schwerwiegender Zwischenfälle und unerwünschter Reaktionen sowie bestimmter technischer Anforderungen an die Kodierung, Verarbeitung, Konservierung und Lagerung und Verteilung von menschlichen Geweben und Zellen, zuletzt geändert durch die Richtlinie (EU) 2015/565 der Kommission vom 8. April 2015; ABl. L 93:43 – 55
- 26.
-
Richtlinie 2015/565/EG der Kommission vom 8. April 2015 zur Änderung der Richtlinie 2006/86/EG hinsichtlich bestimmter technischer Vorschriften für die Kodierung menschlicher Gewebe und Zellen, Text von Bedeutung für den EWR; ABl. L 93: 43 – 55
- 27.
-
Richtlinie 2015/566/EG der Kommission vom 8. April 2015 zur Durchführung der Richtlinie 2004/23/EG hinsichtlich der Verfahren zur Prüfung der Gleichwertigkeit von Qualitäts- und Sicherheitsstandards bei eingeführten Geweben und Zellen, Text von Bedeutung für den EWR; ABl. L 93: 56 – 68
- 28.
-
Richtlinien zur Gewinnung von Blut und Blutbestandteilen und zur Anwendung von Blutprodukten (Hämotherapie) gemäß den §§ 12 und 18 TFG (Gesamtnovelle 2005); BAnz. Nr. 209a vom 5. November 2005 in Verbindung mit der ersten Richtlinienanpassung 2007; BAnz. Nr. 92 vom 19. Mai 2007 und der zweiten Richtlinienanpassung 2010, BAnz. Nr. 101a vom 9. Juli 2010
- 29.
-
Schroeter J, Maier P, Bednarz J, Blüthner K, Quenzel M, Pruss A, Reinhard T: Procedural guidelines. Good tissue practice for cornea banks. Ophthalmologe 2009; 106(3):265 – 74
- 30.
-
Schroeter J, Rieck P: Endothelial evaluation in the cornea bank. Dev Ophthalmol 2009; 43:47 – 62
- 31.
-
The European Pharmacopoeia Monograph of Bovine Serum 5.4. 2006:2262
- 32.
-
Toniolo M, Camposampiero D, Griffoni C, Jones GL: Quality Management in European eye banks. Dev Ophthalmol 2009; 43:70 – 86
- 33.
-
von Auer F: Das Gewebegesetz – Hintergründe und Konsequenzen. Transfusion Medicine and Hemotherapy 2008; 35:407 – 13
- 34.
-
Validierungsempfehlungen des Paul-Ehrlich-Instituts bei der Testung postmortaler Blutproben, http://www.pei.de/DE/infos/pu/genehmigungen/gewebe-21-a-amg/leichenblut/validierungsempfehlungen-testsysteme-node.html
- 35.
-
Wilhelm FW, Duncker GIW, Bredehorn T: Augenbanken, Walter de Gruyter Verlag 2002
17.3 Abkürzungsverzeichnis
| AMG | Gesetz über den Verkehr mit Arzneimitteln (Arzneimittelgesetz) |
| AMWHV | Verordnung über die Anwendung der Guten Herstellungspraxis bei der Herstellung von Arzneimitteln und Wirkstoffen und über die Anwendung der „Guten Fachlichen Praxis“ bei der Herstellung von Produkten menschlicher Herkunft (Arzneimittel- und Wirkstoffherstellungsverordnung) |
| HIV | Human immunodeficiency virus |
| HTLV | Humanes T-lymphotropes Virus |
| MEM | Minimum Essential Medium Eagle |
| SAE | Systemic adverse events |
| SAR | Severe adverse reactions |
| TPG | Gesetz über Spende, Entnahme und Übertragung von Organen und Geweben (Transplantationsgesetz) |
| TPG-GewV | Verordnung über die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung nach dem Transplantationsgesetz (TPG-Gewebeverordnung) |
17.4 Angehörte Sachverständige
Arbeitsgemeinschaft der Obersten Landesgesundheitsbehörden (AOLG)
Bundesministerium für Gesundheit (BMG)
Deutsche Gesellschaft für Hygiene und Mikrobiologie e. V. (DGHM)
Deutsche Gesellschaft für Pathologie e. V. (DGP)
Deutsche Gesellschaft für Rechtsmedizin e. V. (DGRM)
Deutsche Gesellschaft für Transfusionsmedizin und Immunhämatologie e. V. (DGTI)
Gesellschaft für Virologie e. V. (GfV)
Deutsche Interdisziplinäre Vereinigung für Intensiv- und Notfallmedizin e. V. (DIVI)
Deutsche Krankenhausgesellschaft (DKG)
Deutsche Ophthalmologische Gesellschaft e. V. (DOG)
GKV-Spitzenverband (Spitzenverband Bund der Krankenkassen gemäß § 217a SGB V)
Prof. Dr. med. Gregor Bein, Federführender des Ständigen Arbeitskreises „Richtlinien zur Gewinnung von Blut und Blutbestandteilen und zur Anwendung von Blutprodukten (Hämotherapie)“ des Wissenschaftlichen Beirats der Bundesärztekammer
Prof. Dr. med. Michael Hallek, Federführender des Arbeitskreises „Richtlinien zur Herstellung und Anwendung von hämatopoetischen Stammzellzubereitungen“ des Wissenschaftlichen Beirats der Bundesärztekammer
Prof. Dr. med. Harald Klüter, Federführender des Ständigen Arbeitskreises „Querschnitts-Leitlinien zur Therapie mit Blutkomponenten und Plasmaderivaten“ des Wissenschaftlichen Beirats der Bundesärztekammer
Prof. Dr. iur. Hans Lilie, Vorsitzender der Ständigen Kommission Organtransplantation der Bundesärztekammer
Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten
17.5 Mitglieder und externe Berater des Arbeitskreises
Mitglieder
Priv.-Doz. Dr. med. Isabelle Bekeredjian-Ding
Leiterin der Abteilung 1 „Mikrobiologie“
Paul-Ehrlich-Institut, Langen
Univ.-Prof. Dr. med. Claus Cursiefen
Geschäftsführender Direktor des Zentrums für Augenheilkunde und
Direktor der Klinik und Poliklinik für Allgemeine Augenheilkunde
Universitätsklinikum Köln
Prof. Dr. med. Christian Drosten
Leiter des Instituts für Virologie
Universitätsklinikum Bonn
Prof. Dr. med. Georg Häcker
Ärztlicher Direktor des Instituts für Medizinische Mikrobiologie und Hygiene
Universitätsklinikum Freiburg
Dr. iur. Marlis Hübner
Leiterin der Rechtsabteilung
Bundesärztekammer, Berlin
Prof. Dr. med. Axel Pruß
Direktor des Instituts für Transfusionsmedizin, Campus Charité Mitte
Charité – Universitätsmedizin Berlin
Prof. Dr. med. Thomas Reinhard (Federführender)
Direktor der Klinik für Augenheilkunde
Universitätsklinikum Freiburg
Dipl. Biol. Katja Rosenbaum
Technische Leitung der LIONS Hornhautbank NRW
Universitätsklinikum Düsseldorf
Dr. med. Jan Schroeter, FEBO
Leiter der Hornhautbank Berlin, Universitätsgewebebank, Institut für Transfusionsmedizin
Charité – Universitätsmedizin Berlin
Prof. Dr. med. Berthold Seitz, FEBO
Direktor der Klinik für Augenheilkunde und Hochschulambulanz
Universitätsklinikum des Saarlandes UKS, Homburg/Saar
Prof. Dr. rer. nat. Ralf Tönjes
Leiter des Fachgebietes „Avitale Gewebezubereitungen, Xenogene Zelltherapeutika“
Paul-Ehrlich-Institut, Langen
Externe Berater
Priv.-Doz. Dr. rer. nat. Johannes Blümel
Leiter des Fachgebietes „Virussicherheit“
Paul-Ehrlich-Institut, Langen
Dr. rer. nat. Holger Lößner
Fachgebiet „Mikrobiologische Sicherheit“
Paul-Ehrlich-Institut, Langen
Dr. rer. nat. Dagmar Schilling-Leiß
Stellvertretende Leiterin des Fachgebietes „Avitale Gewebezubereitungen, Xenogene
Zelltherapeutika“
Paul-Ehrlich-Institut, Langen
Dr. med. vet. Utta Schurig
Fachgebiet „Mikrobiologische Sicherheit“
Paul-Ehrlich-Institut, Langen
Dr. rer. nat. Albert Stühler
Fachgebiet „Virussicherheit“
Paul-Ehrlich-Institut, Langen
Geschäftsführung
Bundesärztekammer
Dezernat 6 – Wissenschaft, Forschung und Ethik
Herbert-Lewin-Platz 1
10623 Berlin
- *
- Soweit im Folgenden Berufs-, Gruppen- und/oder Personenbezeichnungen Verwendung finden, ist stets auch die jeweils weibliche Form erfasst. Ausschließlich aus Gründen der Lesbarkeit wird in diesen Fällen auf die gleichzeitige Verwendung männlicher und weiblicher Sprachformen verzichtet.